Electronic Supporting Information. Synthesis and Reactivity of 18 F-Labeled α,α-difluoro-α-aryloxyacetic Acids
|
|
|
- Σκύλλα Γιαννακόπουλος
- 6 χρόνια πριν
- Προβολές:
Transcript
1 Electronic Supporting Information Synthesis and Reactivity of 18 F-Labeled α,α-difluoro-α-aryloxyacetic Acids Tanatorn Khotavivattana,, Samuel Calderwood,, Stefan Verhoog, Lukas Pfeifer, Sean Preshlock, Neil Vasdev,,# Thomas L. Collier,,#,^ and Véronique Gouverneur*, Chemistry Research Laboratory, University of xford, Mansfield Road, xford X1 3TA, U.K. Division of Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital, 55 Fruit Street, Boston, USA. # Department of Radiology, Harvard Medical School, 55 Fruit Street, Boston, USA. ^Advion BioSystems, 10 Brown Road, Suite 101, Ithaca, New York, USA. 1
2 Table of Contents 1. General Experimental Information 3 2. General Experimental Procedures and Characterisation Data Aryl-CFHC2Et Aryl-CFBrC2Et (1) Aryl-CF2C2Et (2) Aryl-CF2C2H (3) Post-Fluorination Reactions ptimisation Table ptimization for the synthesis of [ 18 F]2a from 1a (S1) ptimization for the synthesis of [ 18 F]3a from 1a (S2) ptimization for the synthesis of [ 18 F]4 from [ 18 F]3a (S3) ptimization for the synthesis of 6 from 3a (S4) General Radiochemical Procedures Radio-HPLC Traces Post-Fluorination Functionalization Specific Activity Calculation References NMR Spectra Aryl-CFHC2Et Aryl-CFBrC2Et (1) Aryl-CF2C2Et (2) Aryl-CF2C2H (3) Post-Fluorination Reactions 98 2
3 1. General Experimental Information All NMR spectra were recorded on Bruker DPX200, DPX250, AV400, AVC500, AVB500 and DRX500 spectrometers. 1 H and 13 C NMR spectra are reported as chemical shifts (δ) in parts per million (ppm) relative to the solvent peak using the Bruker internal referencing procedure (edlock). 19 F NMR spectra are referenced relative to CFCl 3 in CDCl 3. Coupling constants (J) are reported in units of hertz (Hz). The following abbreviations are used to describe multiplicities s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) and br s (broad singlet). High resolution mass spectra (HRMS, m/z) were recorded on a Bruker MicroTF spectrometer using positive electrospray ionization (ESI + ) or on a Micromass GCT spectrometer using filed ionization (FI + ) or chemical ionization (CI + ). Infrared spectra were recorded either as the neat compound or in a solution using a Bruker Tensor 27 FT-IR spectrometer. Absorptions are reported in wavenumbers (cm -1 ) and only peaks of interest are reported. ptical rotations were measured on a PerkinElmer Polarimeter model 341 Specific rotations are reported in 10-1 deg cm 2 g -1 and concentrations in g/100 ml. Melting points of solids were measured on a Griffin apparatus and are uncorrected. IUPAC names were obtained using the ACD/I- Lab service. Solvents were purchased from Fisher, Rathburn or Sigma-Aldrich. When dry solvents were required they were purified by expression through an activated alumina column built according to the procedures described by Pangborn and Grubbs. 1 Chemicals were purchased from Acros, Alfa Aesar, Fisher, Fluorochem, Sigma-Aldrich and used as received. Reactions were monitored by thin layer chromatography (TLC) carried out on Merck Kiesegel 60 F254 plates, silica gel column chromatography was performed over Merck silica gel C60 (40-60 μm). 3
4 2. General Experimental Procedures and Characterisation Data Aryl-CFBrC 2Et precursors (1), Aryl-CF 2C 2Et references (2), Aryl-CF 2C 2H references (3) and other fluorinated references were purchased from commercial sources, or synthesised based on procedures detailed below. General Procedure A: Preparation of Aryl-CFHC 2Et from Phenols 2 To a suspension of K 2C 3 (25 mmol) in a solution of an appropriate phenol (10 mmol) in DMF (20 ml) was slowly added ethyl bromofluoroacetate (15 mmol) at room temperature. The reaction mixture was stirred at room temperature for 16 hours under nitrogen atmosphere. The reaction mixture was poured on to water, extracted with Et 2 (3 100 ml), dried over MgS 4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography. General Procedure B: Preparation of Aryl-CFBrC 2Et (1) from Aryl-CFHC 2Et To a solution of Aryl-CHFC 2Et (4.0 mmol) in CCl 4 (30 ml) was added N-bromosuccinimide (2.4 mmol). The reaction was allowed to stir for 16 hours at room temperature under UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column chromatography. General Procedure C: Preparation of Aryl-CF 2C 2Et (2) from Phenols 2 To a suspension of K 2C 3 (25 mmol) in a solution of an appropriate phenol (10 mmol) in DMF (20 ml) was slowly added ethyl bromodifluoroacetate (15 mmol) at room temperature. The reaction mixture was stirred at room temperature for 16 hours under nitrogen atmosphere. The reaction mixture was poured on to water, extracted with Et 2 (3 100 ml), dried over MgS 4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography. General Procedure D: Preparation of Aryl-CF 2C 2H (3) from Aryl-CF 2C 2Et (2) 2 To a solution of an Aryl-CF 2C 2Et (5.0 mmol) in Et 2 (25 ml) was added 5 M aq. NaH (25.0 mmol). The reaction mixture was stirred at room temperature for 3 hours before diluting with Et 2. The reaction mixture was extracted with sat. aq. NaHC 3 (3 x 20 ml). The combined aqueous layers were acidified to ph 1 and extracted with Et 2 (3 x 20 ml), then the combined organic phases were washed with brine, dried over MgS 4 and concentrated in vacuo. Products were purified by washing with n-pentane. 2.1 Aryl-CFHC2Et Ethyl (4-tert-butylphenoxy)(fluoro)acetate The title compound was prepared following general procedure A using potassium carbonate (13.8 g, mmol), 4-tert-butylphenol (6.00 g, 40.0 mmol), ethyl bromofluoroacetate (7.10 ml, 60.0 mmol) and DMF (80 ml). The crude product was purified by silica gel column chromatography (eluent: 10 % Et 2 in petroleum ether) to provide the title compound (7.92 g, 78 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.37 (dt, J 1 = 2.6 Hz, J 2 = 8.8 Hz, 2H), 7.08 (d, J = 8.8 Hz, 2H), 5.95 (d, J = 60.1 Hz, 1H), 4.37 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H), 1.32 (s, 9H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.8 Hz), (d, J = 3.2 Hz), 147.3, 126.6, 117.0, (d, J = Hz), 62.5, 34.3, 31.4, 13.9; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 60.7 Hz); IR: ν 2964, 1769, 1608, 1511, 1364, 1216, 1185, 1097, 1014, 965, 833; HRMS (ESI) for C 14H 19FNa 3 [M+Na] + requires found Data consistent with literature values. 2 4
5 Ethyl (biphenyl-4-yloxy)(fluoro)acetate The title compound was prepared following general procedure A using potassium carbonate (3.46 g, 25.0 mmol), 4-phenylphenol (1.70 g, 10.0 mmol), ethyl bromofluoroacetate (1.77 ml, 15.0 mmol) and DMF (20 ml). The crude product was purified by silica gel column chromatography (eluent: 15 % Et 2 in petroleum ether) to provide the title compound (2.57 g, 94 % yield) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 4H), (m, 2H), (m, 1H), 7.24 (d, J = 8.6 Hz, 2H), 6.04 (d, J = 59.9 Hz, 1H), 4.40 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), 155.1, 140.1, 137.5, 128.9, 128.4, 127.2, 126.8, 117.7, (d, J = Hz), 62.5, 13.9; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 60.7 Hz); IR: ν 1768, 1608, 1518, 1486, 1217, 1103, 1007, 838; HRMS (ESI) for C 16H 15FNa 3 [M+Na] + requires found ; Mp C. Data consistent with literature values. 2 Ethyl fluoro(phenoxy)acetate The title compound was prepared following general procedure A using potassium carbonate (6.91 g, 50.0 mmol), phenol (1.88 g, 20.0 mmol), ethyl bromofluoroacetate (3.55 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 20 % Et 2 in n-pentane) to provide the title compound (1.28 g, 32 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2H), (m, 3H), 5.98 (d, J = 59.9 Hz, 1H), 4.37 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), (d, J = 3.2 Hz), 129.8, 124.4, 117.4, (d, J = Hz), 62.5, 14.0; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 60.7 Hz). Data consistent with literature values. 3 Ethyl (4-bromophenoxy)(fluoro)acetate The title compound was prepared following general procedure A using potassium carbonate (10.4 g, 75.0 mmol), 4-bromophenol (5.19 g, 30.0 mmol), ethyl bromofluoroacetate (5.32 ml, 45.0 mmol) and DMF (60 ml). The crude product was purified by silica gel column chromatography (eluent: 10 % Et 2 in n-pentane) to provide the title compound (4.46 g, 54 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.47 (dt, J 1 = 2.7 Hz, J 2 = 9.1 Hz, 2H), 7.03 (dt, J 1 = 2.4 Hz, J 2 = 9.1 Hz, 2H), 5.91 (d, J = 59.7 Hz, 1H), 4.37 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), (d, J = 3.2 Hz), 132.8, (d, J = 1.6 Hz), 117.2, (d, J = Hz), 62.7, 14.0; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 60.7 Hz); IR: ν 1765, 1584, 1486, 1384, 1211, 1175, 1099, 1071, 1008, 965, 825; HRMS (ESI) for C 10H 10BrFNa 3 [M+Na] + requires found Data consistent with literature values. 2 5
6 Ethyl (3-bromophenoxy)(fluoro)acetate The title compound was prepared following general procedure A using potassium carbonate (10.4 g, 75.0 mmol), 3-bromophenol (5.19 g, 30.0 mmol), ethyl bromofluoroacetate (5.32 ml, 45.0 mmol) and DMF (60 ml). The crude product was purified by silica gel column chromatography (eluent: 10 % Et 2 in n-pentane) to provide the title compound (6.88 g, 83 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2H), (m, 1H), (m, 1H), 5.94 (q, J = 59.2, 1H), 4.37 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), (d, J = 2.4 Hz), 130.9, 127.6, 122.8, 121.0, 116.1, (d, J = Hz), 62.7, 13.9; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 59.0 Hz); IR: ν 1761, 1582, 1474, 1382, 1209, 1102, 1023, 965, 884; HRMS (ESI) for C 10H 10BrFNa 3 [M+Na] + requires found Data consistent with literature values. 2 Ethyl (2-bromophenoxy)(fluoro)acetate The title compound was prepared following general procedure A using potassium carbonate (6.91 g, 50.0 mmol), 2-bromophenol (3.46 g, 20.0 mmol), ethyl bromofluoroacetate (3.55 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 20 % Et 2 in n-pentane) to provide the title compound (4.68 g, 85 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.60 (dd, J 1 = 1.5 Hz, J 2 = 7.8 Hz, 1H), (m, 1H), 7.23 (dt, J 1 = 1.4 Hz, J 2 = 8.1 Hz, 1H), (m, 1H), 5.91 (d, J = 59.2 Hz, 1H), (m, 2H), 1.38 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), (d, J = 3.2 Hz), 133.8, 128.8, 126.0, 119.2, (d, J = 1.6 Hz), (d, J = Hz), 62.6, 14.0; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 59.0 Hz); IR: ν 1766, 1579, 1477, 1445, 1384, 1218, 1134, 1095, 1028, 965, 857; HRMS (EI/FI) for C 10H 10BrF 3 [M] + requires found Data consistent with literature values. 2 Ethyl 2-fluoro-2-(2-fluorophenoxy)acetate The title compound was prepared following general procedure A using K 2C 3 (6.91 g, 50.0 mmol), 2- fluorophenol (1.79 ml, 20.0 mmol), ethyl bromofluoroacetate (3.55 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 0-10% Et 2 in n-pentane) to provide the title compound (2.68 g, 12.4 mmol, 62%) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 1 H), (m, 3 H), 6.02 (d, J = 59.2 Hz, 1 H), 4.46 (q, J = 7.1 Hz, 2 H), 1.46 (t, J = 7.2 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), (dd, J = 1.6, Hz), (dd, J = 2.4, 11.1 Hz), (d, J = 7.2 Hz), (d, J = 4.0 Hz), 120.5, (d, J = 18.3 Hz), (dd, J = 1.6, Hz), 62.6, 13.9; ( 19 F NMR, 377 MHz, CDCl 3): δ = (dd, J = 3.4, 59.5 Hz), (m); IR (neat): ν 3076, 2987, 2942, 2906, 2365, 2329, 1764, 1604, 1503, 1459, 1415, 1377, 1352, 1303, 1261, 1229, 1193, 1122, 1090, 1058, 1017, 961, 858, 818, 754, 676; HRMS (ESI) for C 10H 10F 2Na 3 [M+Na] + requires found
7 tert-butyl 4-hydroxybenzoate To a solution of 4-hydroxybenzoic acid (6.91 g, 50.0 mmol), 4-DMAP (244 mg, 2.00 mmol) and tertbutanol (150 ml) in THF (220 ml) at rt was slowly added DCC (11.3 g, 55.0 mmol) dissolved in THF (80 ml). The resulting solution was stirred at rt for 23 h. The formed solid was filtered off and the filtrate was washed with aq. Na2C3 (0.30 M), dried over MgS4 and concentrated in vacuo. The obtained crude product was purified by silica gel column chromatography (eluent: 0-15% Et2 in n- pentane) to give the title compound (5.50 g, 28.3 mmol, 57%) as a white solid. ( 1 H NMR, 400 MHz, CDCl3): δ = (m, 2 H), 7.48 (br s, 1 H), (m, 2 H), 1.60 (s, 9 H); ( 13 C NMR, 101 MHz, CDCl3): δ = 166.7, 160.3, 131.7, 123.6, 115.2, 81.3, Data consistent with literature values. 11 tert-butyl 4-(2-ethoxy-1-fluoro-2-oxoethoxy)benzoate The title compound was prepared following general procedure A using K 2C 3 (5.18 g, 37.5 mmol), tertbutyl 4-hydroxybenzoate (2.91 g, 15.0 mmol), ethyl bromofluoroacetate (2.66 ml, 22.5 mmol) and DMF (30 ml). The crude product was purified by silica gel column chromatography (eluent: 0-12% Et 2 in n-pentane) to provide the title compound (3.41 g, 11.4 mmol, 76%) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): = (m, 2 H), (m, 2 H), 6.02 (d, J = 59.2 Hz, 1 H), 4.35 (q, J = 7.1 Hz, 2 H), 1.57 (s, 9 H), 1.35 (t, J = 7.2 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): = 164.8, (d, J = 31.0 Hz), (d, J = 2.4 Hz), 131.5, 128.0, 116.5, (d, J = Hz), 81.0, 62.7, 28.1, 13.9; ( 19 F NMR, 377 MHz, CDCl 3): = (d, J = 59.5 Hz); IR (neat): ν 2980, 2934, 2903, 2874, 2364, 2329, 1766, 1709, 1604, 1508, 1467, 1452, 1420, 1368, 1294, 1217, 1161, 1102, 1057, 1014, 959, 852, 770, 695; HRMS (ESI) for C 15H 19FNa 5 [M+Na] + requires found Ethyl (4-benzoylphenoxy)(fluoro)acetate The title compound was prepared following general procedure A using potassium carbonate (6.91 g, 50.0 mmol), 4-hydroxybenzophenone (3.96 g, 20.0 mmol), ethyl bromofluoroacetate (3.55 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 30 % Et 2 in n-pentane) to provide the title compound (5.50 g, 91 % yield) as a pale yellow oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.85 (dt, J 1 = 2.3 Hz, J 2 = 8.8 Hz, 2H), (m, 2H), 7.59 (tt, J 1 = 1.9 Hz, J 2 = 7.5 Hz, 1H), (m, 2H), 7.21 (d, J = 8.6 Hz, 2H), 6.08 (d, J = 59.2 Hz, 1H), 4.38 (q, J = 7.2 Hz, 2H), 1.38 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 195.3, (d, J = 31.0 Hz), (d, J = 2.4 Hz), 137.5, 133.5, 132.4, 132.3, 129.8, 128.3, 116.6, (d, J = Hz), 62.8, 13.9; ( 19 F NMR, 7
8 377 MHz, CDCl 3): δ = (d, J = 59.0 Hz); IR: ν 1766, 1656, 1600, 1505, 1447, 1278, 1218, 1174, 1151, 1096, 1025, 939, 924, 848; HRMS (ESI) for C 17H 16FNa 3 [M+H] + requires found Data consistent with literature values. 2 Ethyl (3-cyanophenoxy)(fluoro)acetate The title compound was prepared following general procedure A using potassium carbonate (6.91 g, 50.0 mmol), 3-cyanophenol (2.38 g, 20.0 mmol), ethyl bromofluoroacetate (3.55 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 25 % Et 2 in n-pentane) to provide the title compound (2.33 g, 52 % yield) as a white crystal. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2H), (m, 2H), 5.97 (q, J = 58.9, 1H), 4.36 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 30.2 Hz), (d, J = 2.4 Hz), 130.8, 128.1, 122.1, 120.8, 117.8, 113.7, (d, J = Hz), 62.8, 13.9; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 59.0 Hz); IR: ν 2234, 1762, 1582, 1482, 1436, 1384, 1228, 1156, 1095, 1018, 966, 859; HRMS (EI/FI) for C 11H 10FN 3 [M] + requires found ; Mp C. Ethyl fluoro[4-(trifluoromethyl)phenoxy]acetate The title compound was prepared following general procedure A using potassium carbonate (6.91 g, 50.0 mmol), 4-(trifluoromethyl)phenol (3.24 g, 20.0 mmol), ethyl bromofluoroacetate (3.55 ml, 30.0 mmol) and DMF (60 ml). The crude product was purified by silica gel column chromatography (eluent: 10 % Et 2 in n-pentane) to provide the title compound (4.74 g, 89 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.63 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 8.8 Hz, 2H), 6.02 (d, J = 59.2 Hz, 1H), 4.38 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), 157.9, (q, J = 4.0 Hz), (q, J = 32.9 Hz), (q, J = Hz), (d, J = 1.6 Hz), (d, J = Hz), 62.8, 13.9; ( 19 F NMR, 377 MHz, CDCl 3): δ = 62.1 (s, 3F), (d, J = 59.0 Hz, 1F); IR: ν 1766, 1616, 1517, 1325, 1222, 1166, 1113, 1065, 1113, 1065, 1014, 966, 840; HRMS (EI/FI) for C 11H 10F 4 3 [M] + requires found Data consistent with literature values. 2 2-(4-Hydroxyphenyl)-1H-isoindole-1,3(2H)-dione To a suspension of phthalic anhydride (5.92 g, 40.0 mmol) in MeH (100 ml) was added 4- aminophenol (4.80 g, 44.0 mmol). The mixture was heated to 70 C over night, before it was cooled to rt and concentrated in vacuo. Recrystallisation from MeH gave the title compound (6.56 g, 27.4 mmol, 69%) as a white solid. ( 1 H NMR, 400 MHz, DMS-d 6): δ = 9.77 (br s, 1 H), (m, 4 H), (m, 2 H), (m, 2 H); ( 13 C NMR, 101 MHz, DMS-d 6): δ = 167.4, 157.3, 134.6, 131.6, 128.8, 123.3, 122.9, Data consistent with literature values. 12 8
9 Ethyl 2-(4-(1,3-dioxoisoindolin-2-yl)phenoxy)-2-fluoroacetate The title compound was prepared following general procedure A using K 2C 3 (5.06 g, 36.6 mmol), 2- (4-hydroxyphenyl)-1H-isoindole-1,3(2H)-dione (3.50 g, 14.6 mmol), ethyl bromofluoroacetate (2.59 ml, 22.0 mmol) and DMF (30 ml). The crude product was purified by silica gel column chromatography (eluent: 20-70% Et 2 in n-pentane) to provide the title compound (2.94 g, 8.55 mmol, 58%) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2 H), (m, 2 H), (m, 2 H), (m, 2 H), 5.98 (d, J = 59.4 Hz, 1 H), 4.36 (q, J = 7.2 Hz, 2 H), 1.36 (t, J = 7.1 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 167.1, (d, J = 31.0 Hz), (d, J = 3.2 Hz), 134.4, 131.5, 128.0, 127.9, 123.7, 118.0, (d, J = Hz), 62.6, 13.9; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 59.5 Hz); IR (neat): ν 3073, 2987, 2365, 2329, 1764, 1711, 1606, 1512, 1464, 1387, 1295, 1222, 1173, 1122, 1089, 1018, 958, 884, 830, 791, 716; MP: 103 C; HRMS (ESI) for C 18H 15FN 5 [M+H] + requires found Aryl-CFBrC2Et (1) Ethyl (4-tert-butylphenoxy)(bromo)fluoroacetate (1a) The title compound was prepared following general procedure B; To a solution of ethyl (4-tertbutylphenoxy)(fluoro)acetate (1.62 g, 6.37 mmol) in CCl 4 (30 ml) was added N-bromosuccinimide (680 mg, 3.82 mmol). The reaction was allowed to stir for 16 hours at room temperature under UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: 2 % Et 2 in n-pentane) to provide the title compound (465 mg, 37 % yield) as a pale yellow oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.41 (dt, J 1 = 2.6 Hz, J 2 = 8.8 Hz, 2H), (m, 2H), 4.45 (q, J = 7.1 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H), 1.34 (s, 9H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), 149.5, 149.4, 126.3, (d, J = 1.6 Hz), (d, J = Hz), 63.9, 34.5, 31.3, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 57.4 (s); IR: ν 2965, 1770, 1510, 1368, 1306, 1207, 1182, 1104, 1015, 919, 859; HRMS (EI/FI) for C 14H 18BrF 3 [M] + requires found Ethyl (biphenyl-4-yloxy)(bromo)fluoroacetate (1b) The title compound was prepared following general procedure B; To a solution of ethyl (biphenyl-4- yloxy)(fluoro)acetate (2.51 g, 9.15 mmol) in CCl 4 (40 ml) was added N-bromosuccinimide (979 mg, 5.5 mmol). The reaction was allowed to stir for 16 hours at 70 C under UV light irradiation. The reaction mixture was cooled to room temperature, then the solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: 5% Et 2 in n-pentane) to provide the title compound (732 mg, 38 % yield) as a white solid. 9
10 ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 4H), (m, 2H), (m, 3H), 4.48 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), 151.1, 140.0, 139.6, 128.8, 128.2, 127.6, 127.1, (d, J = 2.4 Hz), (d, J = Hz), 64.1, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 57.6 (s); IR: ν 1515, 1485, 1371, 1305, 1204, 1171, 1100, 1008, 919, 855; HRMS (EI/FI) for C 16H 14BrF 3 [M] + requires found ; Mp C. Ethyl bromo(phenoxy)fluoroacetate (1c) The title compound was prepared following general procedure B; to a solution of ethyl fluoro(phenoxy)acetate (793 mg, 4.0 mmol) in CCl 4 (20 ml) was added N-bromosuccinimide (427 mg, 2.4 mmol). The reaction was allowed to stir for 16 hours at 70 C under UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: 5 % Et 2 in n-pentane) to provide the title compound (208 mg, 31 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2H), (m, 3H), 4.37 (q, J = 7.1 Hz, 2H), 1.34 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), 151.8, 129.5, 126.5, 121.8, (d, J = Hz), 64.0, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 57.5 (s); IR: ν 1767, 1592, 1488, 1371, 1305, 1193, 1164, 1098, 1013, 924, 848; HRMS (EI/FI) for C 10H 10BrF 3 [M] + requires found Ethyl (4-bromophenoxy)(bromo)fluoroacetate (1d) The title compound was prepared following general procedure B; to a solution of ethyl (4- bromophenoxy)(fluoro)acetate (1.11 g, 4.0 mmol) in CCl 4 (20 ml) was added N-bromosuccinimide (427 mg, 2.4 mmol). The reaction was allowed to stir for 16 hours at 70 C under UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: 3 % Et 2 in n-pentane) to provide the title compound (299 mg, 35 % yield) as a pale yellow oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.53 (dt, J 1 = 2.7 Hz, J 2 = 9.1 Hz, 2H), (m, 2H), 4.45 (q, J = 7.1 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 30.2 Hz), 150.7, 132.6, 123.6, 119.9, (d, J = Hz), 64.1, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 58.4 (s); IR: ν 3301, 1760, 1666, 1609, 1543, 1509, 1411, 1372, 1312, 1209, 1103, 1016, 835; HRMS (EI/FI) for C 10H 9Br 2F 3 [M] + requires found Ethyl (3-bromophenoxy)(bromo)fluoroacetate (1e) The title compound was prepared following general procedure B; to a solution of ethyl (3- bromophenoxy)(fluoro)acetate (1.11 g, 4.0 mmol) in CCl 4 (20 ml) was added N-bromosuccinimide (427 mg, 2.4 mmol). The reaction was allowed to stir for 16 hours at 70 C under UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column 10
11 chromatography (eluent: 5 % Et 2 in n-pentane) to provide the title compound (221 mg, 26 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2H), (m, 2H), 4.37 (q, J = 7.1 Hz, 2H), 1.34 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), 152.1, 130.6, 129.8, (d, J = 2.4 Hz), 122.4, 120.5, (d, J = Hz), 64.1, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 58.4 (s); IR: ν 2971, 1766, 1582, 1471, 1371, 1304, 1199, 1148, 1100, 1013, 924, 874; HRMS (EI/FI) for C 10H 9Br 2F 3 [M] + requires found Ethyl (2-bromophenoxy)(bromo)fluoroacetate (1f) The title compound was prepared following general procedure B; to a solution of ethyl (2- bromophenoxy)(fluoro)acetate (1.11 g, 4.0 mmol) in CCl 4 (20 ml) was added N-bromosuccinimide (427 mg, 2.4 mmol). The reaction was allowed to stir for 16 hours at 70 C under UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: 5 % Et 2 in n-pentane) to provide the title compound (195 mg, 23 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.65 (dd, J 1 = 1.5 Hz, J 2 = 8.1 Hz, 1H), 7.47 (dt, J 1 = 1.7 Hz, J 2 = 8.3 Hz, 1H), 7.36 (td, J 1 = 1.5 Hz, J 2 = 7.8 Hz, 1H), 7.18 (td, J 1 = 1.5 Hz, J 2 = 7.7 Hz, 1H), 4.46 (qd, J 1 = 1.5 Hz, J 2 = 7.1 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.8 Hz), 149.2, 133.9, 128.4, 127.6, (d, J = 2.4 Hz), (d, J = 1.6 Hz), (d, J =303.6 Hz), 64.1, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 56.7 (s); IR: ν 1767, 1473, 1445, 1371, 1305, 1212, 1147, 1099, 1047, 1031, 1012, 923, 850; HRMS (EI/FI) for C 10H 9Br 2F 3 [M] + requires found Ethyl 2-bromo-2-fluoro-2-(2-fluorophenoxy)acetate (1g) The title compound was prepared following general procedure B using NBS (997 mg, 5.60 mmol), ethyl fluoro(2-fluorophenoxy)acetate (865 mg, 4.00 mmol) and CCl 4 (20 ml). The crude product was purified by silica gel column chromatography (eluent: 0-5% Et 2 in n-pentane) to provide the title compound (235 mg, 0.80 mmol, 20%) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.51 (tt, J = 1.8, 7.9 Hz, 1 H), (m, 1 H), (m, 2 H), 4.55 (q, J = 7.3 Hz, 2 H), 1.51 (t, J = 7.2 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 30.2 Hz), (d, J = Hz), (d, J = 11.9 Hz), (d, J = 7.2 Hz), (d, J = 1.6 Hz), (d, J = 4.0 Hz), (d, J = 18.3 Hz), (d, J = Hz), 64.1, 13.7; ( 19 F NMR, 377 MHz, CDCl 3): δ = (d, J = 10.3 Hz), (m); IR (neat): ν 3076, 2988, 2905, 2364, 2329, 1841, 1768, 1601, 1500, 1458, 1403, 1374, 1303, 1258, 1189, 1148, 1096, 1012, 951, 919, 859, 820, 757, 699; HRMS (ESI) for C 10H 9BrF 2Na 3 [M+Na] + requires found
12 tert-butyl 4-(1-bromo-2-ethoxy-1-fluoro-2-oxoethoxy)benzoate (1h) The title compound was prepared following general procedure B using NBS (854 mg, 4.80 mmol), tertbutyl 4-(2-ethoxy-1-fluoro-2-oxoethoxy)benzoate (1.19 g, 4.00 mmol) and CCl 4 (20 ml). The crude product was purified by silica gel column chromatography (eluent: 0-3% Et 2 in n-pentane) to provide the title compound (394 mg, 1.04 mmol, 26%) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2 H), (m, 2 H), 4.44 (dq, J = 0.7, 7.2 Hz, 2 H), 1.59 (s, 9 H), 1.40 (t, J = 7.2 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 164.6, (d, J = 30.2 Hz), 154.7, 131.0, 130.1, (d, J = 2.4 Hz), (d, J = Hz), 81.3, 64.1, 28.1, 13.7; ( 19 F NMR, 377 MHz, CDCl 3): δ = (s); IR (neat): ν 3079, 2979, 2934, 2902, 2872, 2367, 2328, 1931, 1770, 1714, 1602, 1505, 1466, 1450, 1404, 1367, 1293, 1250, 1204, 1157, 1105, 1012, 922, 853, 775, 736, 693, 649; HRMS not found using ESI, EI, FI. Ethyl (4-benzoylphenoxy)(bromo)fluoroacetate (1i) The title compound was prepared following general procedure B; to a solution of ethyl (4- benzoylphenoxy)(fluoro)acetate (1.21 g, 4.0 mmol) in CCl 4 (20 ml) was added N-bromosuccinimide (427 mg, 2.4 mmol). The reaction was allowed to stir for 16 hours at 70 C under UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: 20 % Et 2 in n-pentane) to provide the title compound (208 mg, 23 % yield) as a pale yellow oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.89 (dt, J 1 = 2.2 Hz, J 2 = 8.8 Hz, 2H), (m, 2H), 7.62 (tt, J 1 = 1.9 Hz, J 2 = 7.3 Hz, 1H), (m, 2H), (m, 2H), 4.47 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 195.4, (d, J = 30.2 Hz), 154.7, 137.3, 135.5, 132.7, 131.8, 130.0, 128.4, (d, J = 2.4 Hz), (d, J = Hz), 64.2, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 59.0 (s); IR: ν 1768, 1660, 1599, 1501, 1447, 1372, 1276, 1203, 1164, 1099, 1014, 921, 856; HRMS (EI/FI) for C 17H 14BrF 4 [M] + requires found Ethyl (3-cyanophenoxy)(bromo)fluoroacetate (1j) The title compound was prepared following general procedure B; to a solution of ethyl (3- cyanophenoxy)(fluoro)acetate (893 mg, 4.0 mmol) in CCl 4 (20 ml) was added N-bromosuccinimide (427 mg, 2.4 mmol). The reaction was allowed to stir for 16 hours at 60 C and UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: 20 % Et 2 in n-pentane) to provide the title compound (113 mg, 16 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 4H), 4.46 (q, J = 7.2 Hz, 2H), 1.42 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 31.0 Hz), 151.7, 130.6, 130.2, (d, J = 1.6 Hz), (d, J 12
13 = 2.4 Hz), 117.5, 113.7, (d, J = Hz), 64.3, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 59.3 (s); IR: ν 2235, 1768, 1581, 1481, 1432, 1372, 1306, 1234, 1158, 1102, 1012, 910, 857; HRMS (EI/FI) for C 11H 9BrFN 3 [M] + requires found Ethyl bromo(fluoro)[4-(trifluoromethyl)phenoxy]acetate (1k) The title compound was prepared following general procedure B; to a solution of ethyl fluoro[4- (trifluoromethyl)phenoxy]acetate (1.06 g, 4.0 mmol) in CCl 4 (20 ml) was added N-bromosuccinimide (427 mg, 2.4 mmol). The reaction was allowed to stir for 16 hours at 60 C under UV light irradiation. The solvent was removed in vacuo. The crude product was purified by silica gel column chromatography (eluent: 5 % Et 2 in n-pentane) to provide the title compound (160 mg, 19 % yield) as a pale yellow oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.69 (d, J = 8.6 Hz, 2H), 7.41 (d, J = 8.6 Hz, 2H), 4.46 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (d, J = 30.2 Hz), 154.1, (q, J = 33.1 Hz), (q, J = 3.7 Hz), (q, J = Hz), (d, J = 2.4 Hz), (d, J =304.4 Hz), 64.2, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 59.2 (s, 1F), 62.3 (s, 3F) ; IR: ν 1771, 1615, 1513, 1325, 1211, 1170, 1128, 1065, 1016, 924, 858; HRMS (EI/FI) for C 11H 9BrF 4 3 [M] + requires found Ethyl 2-bromo-2-(4-(1,3-dioxoisoindolin-2-yl)phenoxy)-2-fluoroacetate (1l) The title compound was prepared following general procedure B using NBS (427 mg, 2.40 mmol), ethyl [4-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)phenoxy](fluoro)acetate (1.37 g, 4.00 mmol) and CCl 4 (20 ml). The crude product was purified by silica gel column chromatography (eluent: 0-50% Et 2 in n- pentane) to provide the title compound (162 mg, 0.38 mmol, 10%) as a white solid alongside some recovered substrate (888 mg, 2.59 mmol). ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2 H), (m, 2 H), (m, 2 H), (m, 2 H), 4.46 (q, J = 7.0 Hz, 2 H), 1.42 (t, J = 7.1 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 167.0, (d, J = 30.2 Hz), 150.7, 134.5, 131.5, 129.9, 127.4, 123.8, (d, J = 1.6 Hz), (d, J = Hz), 64.1, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = (s); IR (neat): ν 3073, 2986, 1768, 1716, 1606, 1506, 1466, 1377, 1301, 1211, 1163, 1108, 1079, 1010, 953, 918, 876, 839, 792, 754, 715, 650; MP: 105 C; HRMS not found using ESI, EI, FI. 2.3 Aryl-CF2C2Et (2) Ethyl (4-tert-butylphenoxy)(difluoro)acetate (2a) The title compound was prepared following general procedure C using potassium carbonate (3.46 g, 25.0 mmol), 4-tert-butylphenol (1.50 g, 10.0 mmol), ethyl bromodifluoroacetate (3.21 ml, 25.0 mmol) 13
14 and DMF (20 ml). The crude product was purified by silica gel column chromatography (eluent: 4 % Et 2 in petroleum ether) to provide the title compound (1.97 g, 72 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.38 (dt, J 1 = 2.6 Hz, J 2 = 8.8 Hz, 2H), 7.15 (d, J = 8.8 Hz, 2H), 4.39 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H), 1.33 (s, 9H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 41.3 Hz), 149.3, 147.1, 126.4, 121.1, (t, J = Hz), 63.5, 34.5, 31.3, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.3 (s); IR: ν 2966, 1777, 1509, 1377, 1341, 1179, 1136, 1108, 1016, 853; HRMS (ESI) for C 14H 18F 2Na 3 [M+Na] + requires found Data consistent with literature values. 2 Ethyl (biphenyl-4-yloxy)(difluoro)acetate (2b) The title compound was prepared following general procedure C using potassium carbonate (3.46 g, 25.0 mmol), 4-phenylphenol (1.70 g, 10.0 mmol), ethyl bromodifluoroacetate (3.21 ml, 25.0 mmol) and DMF (20 ml). The crude product was purified by silica gel column chromatography (eluent: 3% EtAc in n-hexane) to provide the title compound (1.60 g, 55 % yield) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 4H), (m, 2H), (m, 1H), 7.31 (d, J = 8.8 Hz, 2H), 4.42 (q, J = 7.1 Hz, 2H), 1.40 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 41.3 Hz), 148.8, , 139.4, 128.8, 128.2, 127.5, 127.1, 121.9, (t, J = Hz), 63.7, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.3 (s); IR: ν 1775, 1515, 1486, 1378, 1341, 1171, 1134, 1008, 854; HRMS (ESI) for C 16H 14F 2Na 3 [M+Na] + requires found ; Mp C. Data consistent with literature values. 2 Ethyl difluoro(phenoxy)acetate (2c) The title compound was prepared following general procedure C using potassium carbonate (6.91 g, 50.0 mmol), phenol (1.88 g, 20.0 mmol), ethyl bromodifluoroacetate (3.85 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 2 % Et 2 in n- pentane) to provide the title compound (1.37 g, 32 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2H), (m, 3H), 4.41 (q, J = 7.2 Hz, 2H), 1.39 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 41.3 Hz), 149.5, 129.6, 126.3, 121.6, (t, J = Hz), 63.6, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.2 (s); IR: ν , 1378, 1341, 1191, 1130, 1005, 858; HRMS (ESI) for C 10H 10F 2Na 3 [M+Na] + requires found Ethyl (4-bromophenoxy)(difluoro)acetate (2d) The title compound was prepared following general procedure C using potassium carbonate (6.91 g, 50.0 mmol), 4-bromophenol (3.46 g, 20.0 mmol), ethyl bromodifluoroacetate (3.85 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 3 % Et 2 in n-pentane) to provide the title compound (2.02 g, 34 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.49 (dt, J 1 = 2.7 Hz, J 2 = 9.1 Hz, 2H), (m, 2H), 4.40 (q, J = 7.2 Hz, 2H), 1.38 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 40.9 Hz), 148.4, 132.7, 14
15 123.4, 119.6, (t, J = Hz), 63.8, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.6 (s); IR: ν 1777, 1485, 1378, 1342, 1198, 1136, 1069, 1012, 848; HRMS (EI/FI) for C 10H 9BrF 2 3 [M] + requires found Ethyl (3-bromophenoxy)(difluoro)acetate (2e) The title compound was prepared following general procedure C using potassium carbonate (6.91 g, 50.0 mmol), 3-bromophenol (3.46 g, 20.0 mmol), ethyl bromodifluoroacetate (3.85 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 1 % Et 2 in n-pentane) to provide the title compound (1.15 g, 20 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2H), (m, 1H), (m, 1H), 4.40 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 40.9 Hz), 149.9, 130.6, 129.6, 125.1, 122.5, 120.3, (t, J = Hz), 63.8, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.6 (s); IR: ν 1776, 1585, 1472, 1341, 1136, 1012, 858; HRMS (EI/FI) for C 10H 9BrF 2 3 [M] + requires found Ethyl (2-bromophenoxy)(difluoro)acetate (2f) The title compound was prepared following general procedure C using potassium carbonate (6.91 g, 50.0 mmol), 2-bromophenol (3.46 g, 20.0 mmol), ethyl bromodifluoroacetate (3.85 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 2 % Et 2 in n-pentane) to provide the title compound (2.11 g, 36 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.63 (dd, J 1 = 1.2 Hz, J 2 = 7.8 Hz, 1H), (m, 2H), (m, 1H), 7.14 (ddd, J 1 = 2.0 Hz, J 2 = 6.9 Hz, J 3 = 7.8 Hz, 1H), 4.42 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 40.9 Hz), 146.8, 133.8, 128.4, 127.5, 123.0, 116.5, (t, J = Hz), 63.8, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.0 (s); IR: ν 1776, 1473, 1446, 1378, 1341, 1212, 1136, 1048, ; HRMS (EI/FI) for C 10H 9BrF 2 3 [M] + requires found Ethyl 2,2-difluoro-2-(2-fluorophenoxy)acetate (2g) The title compound was prepared following general procedure C using K 2C 3 (6.91 g, 50.0 mmol), 2- fluorophenol (1.79 g, 20.0 mmol), ethyl bromodifluoroacetate (3.85 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 0-7% Et 2 in n-pentane) to provide the title compound (2.25 g, 9.62 mmol, 48%) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.33 (qt, J = 1.3, 7.9 Hz, 1 H), (m, 1 H), (m, 2 H), 4.42 (q, J = 7.2 Hz, 2 H), 1.40 (t, J = 7.2 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 40.5 Hz), (d, J = Hz), (td, J = 2.2, 12.3 Hz), (d, J = 7.2 Hz), 124.4, (d, J = 4.0 Hz), (d, J = 18.3 Hz), (t, J = Hz), 63.7, 13.5; ( 19 F NMR, 377 MHz, CDCl 3): δ = (s, 2F), (m, 1F); IR (neat): ν 2990, 2943, 2364, 2329, 1777, 1603, 1502, 1459, 1410, 1382, 1339, 15
16 1261, 1187, 1133, 1090, 1013, 941, 894, 851, 792, 751, 695; HRMS (ESI) for C 10H 9F 3Na 3 [M+Na] + requires found tert-butyl 4-(2-ethoxy-1,1-difluoro-2-oxoethoxy)benzoate (2h) The title compound was prepared following general procedure C using K 2C 3 (3.46 g, 25.0 mmol), tertbutyl 4-hydroxybenzoate (1.94 g, 10.0 mmol), ethyl bromodifluoroacetate (1.92 ml, 15.0 mmol) and DMF (20 ml). The crude product was purified by silica gel column chromatography (eluent: 0-15% Et 2 in n-pentane) to provide the title compound (965 mg, 3.05 mmol, 31%) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2 H), (m, 2 H), 4.38 (q, J = 7.1 Hz, 2 H), 1.58 (s, 9 H), 1.36 (t, J = 7.1 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 164.7, (t, J = 40.9 Hz); (t, J = 1.6 Hz), 131.1, 129.9, 120.7, (t, J = Hz), 81.3, 63.8, 28.1, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = (s); IR (neat): ν 2981, 2935, 2904, 2876, 1778, 1714, 1604, 1506, 1466, 1451, 1407, 1375, 1338, 1294, 1253, 1207, 1158, 1106, 1011, 962, 855, 757, 696; HRMS (ESI) for C 15H 18F 2Na 5 [M+Na] + requires found Ethyl (4-benzoylphenoxy)(difluoro)acetate (2i) The title compound was prepared following general procedure C using potassium carbonate (6.91 g, 50.0 mmol), 4-hydroxybenzophenone (3.96 g, 20.0 mmol), ethyl bromodifluoroacetate (3.85 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 5 % Et 2 in n-pentane) to provide the title compound (2.26 g, 35 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.84 (dt, J 1 = 2.2 Hz, J 2 = 8.8 Hz, 2H), (m, 2H), (m, 1H), 7.49 (t, J = 7.6 Hz, 2H), 7.33 (d, J = 8.8 Hz, 2H), 4.41 (q, J = 7.2 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 195.3, (t, J = 41.0 Hz), 152.6, 137.2, 135.3, 132.6, 131.8, 129.9, 128.3, 120.8, (d, J = Hz), 63.8, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.5 (s); IR: ν 1776, 1660, 1599, 1502, 1448, 1378, 1276, 1167, 1014, 938, 924, 855; HRMS (ESI) for C 17H 15F 2 4 [M+H] + requires found Data consistent with literature values. 2 Ethyl (3-cyanophenoxy)(difluoro)acetate (2j) The title compound was prepared following general procedure C using potassium carbonate (6.91 g, 50.0 mmol), 3-cyanophenol (2.38 g, 20.0 mmol), ethyl bromodifluoroacetate (3.85 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 20 % Et 2 in n-pentane) to provide the title compound (197 mg, 4 % yield) as a pale yellow oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 1H), (m, 3H), 4.41 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 40.1 Hz), 149.5, 130.7, 130.0, 126.3, 125.1, 16
17 117.5, 113.8, (t, J = Hz), 64.0, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.8 (s); IR: ν 2236, 1775, 1582, 1483, 1432, 1378, 1342, 1238, 1148, 1010, 854; HRMS (EI/FI) for C 11H 9F 2N 3 [M] + requires found Ethyl difluoro[4-(trifluoromethyl)phenoxy]acetate (2k) The title compound was prepared following general procedure C using potassium carbonate (6.91 g, 50.0 mmol), 4-(trifluoromethyl)phenol (3.24 g, 20.0 mmol), ethyl bromodifluoroacetate (3.85 ml, 30.0 mmol) and DMF (40 ml). The crude product was purified by silica gel column chromatography (eluent: 1 % Et 2 in n-pentane) to provide the title compound (1.36 g, 24 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.66 (d, J = 8.6 Hz, 2H), 7.34 (d, J = 8.6 Hz, 2H), 4.41 (q, J = 7.2 Hz, 2H), 1.38 (t, J = 7.2 Hz, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 40.5 Hz), , (q, J = 33.1 Hz), (q, J = 3.7 Hz), (q, J = Hz), 121.6, (t, J =274.2 Hz), 63.4, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = 62.4 (s, 3F), 76.7 (s, 2F); IR: ν 1779, 1617, 1515, 1379, 1325, 1212, 1126, 1065, 1017, 857; HRMS (EI/FI) for C 11H 9F 5 3 [M] + requires found Ethyl 2-(4-(1,3-dioxoisoindolin-2-yl)phenoxy)-2,2-difluoroacetate (2l) The title compound was prepared following general procedure C using K 2C 3 (2.76 g, 20.0 mmol), 2- (4-hydroxyphenyl)-1H-isoindole-1,3(2H)-dione (1.91 g, 8.0 mmol), ethyl bromodifluoroacetate (1.54 ml, 12.0 mmol) and DMF (16 ml). The crude product was purified by silica gel column chromatography (eluent: 20-70% Et 2 in n-pentane) to provide the title compound (294 mg, 0.81 mmol, 10%) as a white, crystalline solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2 H), (m, 2 H), (m, J = 8.9 Hz, 2 H), (m, J = 8.8 Hz, 2 H), 4.41 (q, J = 7.2 Hz, 2 H), 1.38 (t, J = 7.1 Hz, 3 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 167.0, (t, J = 41.1 Hz), (t, J = 2.2 Hz), 134.5, 131.6, 129.7, 127.6, 123.8, 122.2, (t, J = Hz), 63.8, 13.8; ( 19 F NMR, 377 MHz, CDCl 3): δ = (s); IR (neat): ν 3113, 3072, 2989, 2941, 2905, 1917, 1857, 1774, 1709, 1605, 1512, 1464, 1391, 1341, 1292, 1221, 1173, 1125, 1080, 1013, 951, 890, 849, 781, 714, 639; MP: C; HRMS (ESI) for C 18H 14F 2N 5 [M+H] + requires found Aryl-CF2C2H (3) 4-tert-Butylphenoxy)(difluoro)acetic acid (3a) The title compound was prepared following general procedure D using ethyl (4-tertbutylphenoxy)(difluoro)acetate (1.63 g, 6.0 mmol), 5 M aq. NaH (6 ml, 30.0 mmol) and MeCN (30 ml); (1.40 g, 96 % yield, white solid). 17
18 ( 1 H NMR, 400 MHz, CDCl 3): δ = 9.90 (brs, 1H), 7.40 (dt, J 1 = 2.6 Hz, J 2 = 9.1 Hz, 2H), 7.17 (d, J = 8.8 Hz, 2H), 1.33 (s, 9H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 42.9 Hz), 149.6, 146.7, 126.6, 121.1, (t, J = Hz), 34.5, 31.3; ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.6 (s); IR: ν 2965, 1765, 1509, 1366, 1176, 1105, 1016, 833, 804; HRMS (ESI) for C 12H 14F 2Na 3 [M+Na] + requires found ; Mp C. Data consistent with literature values. 2 (Biphenyl-4-yloxy)(difluoro)acetic acid (3b) The title compound was prepared following general procedure D using ethyl (biphenyl-4- yloxy)(difluoro)acetate (640 mg, 2.19 mmol), 4 M aq. NaH (5.5 ml, 22.0 mmol) and MeCN (25 ml); (588 mg, 100 % yield, grey solid). ( 1 H NMR, 400 MHz, CDCl 3): δ = 9.77 (brs, 1H), (m, 4H), (m, 2H), (m, 1H), 7.33 (d, J = 8.6 Hz, 2H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 42.9 Hz), 148.5, 139.9, 139.7, 128.9, 128.4, 127.6, 127.1, 121.9, (t, J = Hz); ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.6 (s); IR: ν 3063, 1776, 1519, 1487, 1452, 1248, 1208, 1190, 1174, 1155, 1101, 1008, 850; HRMS (ESI) for C 14H 9F 2 3 [M- H] - requires found ; Mp C. Data consistent with literature values. 2 Difluoro(phenoxy)acetic acid (3c) The title compound was prepared following general procedure D using ethyl difluoro(phenoxy)acetate (865 mg, 4.0 mmol), 5 M aq. NaH (4 ml, 20.0 mmol) and MeCN (20 ml); (742 mg, 99 % yield, yellow oil). ( 1 H NMR, 400 MHz, CDCl 3): δ = 8.81 (brs, 1H), (m, 2H), (m, 3H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 42.9 Hz), 149.1, 129.7, 126.6, 121.6, (t, J = Hz); ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.7 (s); IR: ν 1762, 1591, 1491, 1189, 1025, 1004, 855; HRMS (EI/FI) for C 8H 6F 2 3 [M] + requires found (4-Bromophenoxy)(difluoro)acetic acid (3d) The title compound was prepared following general procedure D using ethyl (4- bromophenoxy)(difluoro)acetate (1.48 g, 5.0 mmol), 5 M aq. NaH (5 ml, 25.0 mmol) and MeCN (25 ml); (880 mg, 76 % yield, white solid). ( 1 H NMR, 400 MHz, CDCl 3): δ = (brs, 1H), 7.52 (dt, J 1 = 2.7 Hz, J 2 = 8.8 Hz, 2H), (m, 2H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 42.1 Hz), 148.1, 132.8, 123.4, 120.0, (t, J = Hz); ( 19 F NMR, 377 MHz, CDCl 3): δ = 77.0 (s); IR: ν 1765, 1484, 1194, 1068, 1013; HRMS (EI/FI) for C 8H 5BrF 2 3 [M] + requires found ; Mp C. 18
19 (3-Bromophenoxy)(difluoro)acetic acid (3e) The title compound was prepared following general procedure D using ethyl (3- bromophenoxy)(difluoro)acetate (590 mg, 2.0 mmol), 5 M aq. NaH (2 ml, 10.0 mmol) and MeCN (10 ml); (382 mg, 71 % yield, pale yellow solid). ( 1 H NMR, 400 MHz, CDCl 3): δ = 8.37 (brs, 1H), (m, 2H), (m, 1H), (m, 1H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 41.3 Hz), 149.5, 130.8, 129.9, 125.1, 122.6, 120.3, (t, J = Hz); ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.9 (s); IR: ν 3574, 1761, 1580, 1462, 1245, 1191, 1142, 1062, 873; HRMS (ESI) for C 8H 4BrF 2 3 [M-H] - requires found ; Mp C. (2-Bromophenoxy)(difluoro)acetic acid (3f) The title compound was prepared following general procedure D using ethyl (2- bromophenoxy)(difluoro)acetate (1.18 g, 4.0 mmol), 5 M aq. NaH (4 ml, 20.0 mmol) and MeCN (20 ml); (1.03 g, 96 % yield, pale yellow oil). ( 1 H NMR, 400 MHz, CDCl 3): δ = (brs, 1H), 7.65 (dd, J 1 = 1.2 Hz, J 2 = 7.8 Hz, 1H), (m, 2H), 7.18 (ddd, J 1 = 2.0 Hz, J 2 = 6.9 Hz, J 3 = 7.8 Hz, 1H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 41.1 Hz), 146.4, 133.9, 128.5, 127.9, 1231, 116.5, (t, J = Hz); ( 19 F NMR, 377 MHz, CDCl 3): δ = 76.5 (s); IR: ν 1766, 1472, 1445, 1149, 1047, 1030, 855; HRMS (EI/FI) for C 8H 5BrF 2 3 [M] + requires found ,2-Difluoro-2-(2-fluorophenoxy)acetic acid (3g) The title compound was prepared following general procedure D using ethyl difluoro(2- fluorophenoxy)acetate (234 mg, 1.00 mmol), aq. NaH (5.0 M; 1.00 ml, 5.00 mmol) and MeCN (5.00 ml). After work-up the title compound (152 mg, 0.74 mmol, 74%) was obtained as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = (br s, 1 H), (m, 1 H), (m, 1 H), (m, 2 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 42.1 Hz), (d, J = Hz), (td, J = 1.6, 12.7 Hz), (d, J = 7.2 Hz), 124.5, 124.5, (d, J = 18.3 Hz), (t, J = Hz); ( 19 F NMR, 377 MHz, CDCl 3): δ = (s), (m, 1 F); IR (neat): ν 3499, 3078, 2896, 2719, 2560, 2362, 2342, 1920, 1763, 1610, 1601, 1501, 1461, 1388, 1260, 1184, 1147, 1098, 1029, 941, 872, 861, 820, 795, 750; HRMS (ESI) for C 8H 4F 3 3 [M-H] - requires found
20 2-(4-(tert-Butoxycarbonyl)phenoxy)-2,2-difluoroacetic acid (3h) The title compound was prepared following general procedure D using tert-butyl 4-(2-ethoxy-1,1- difluoro-2-oxoethoxy)benzoate (125 mg, 0.40 mmol), aq. NaH (5.0 M; 400 µl, 2.00 mmol) and MeCN (2.00 ml). After work-up the title compound (85 mg, 0.29 mmol, 74%) was obtained as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 9.23 (br s, 1 H), (m, 2 H), (m, 2 H), 1.52 (s, 9 H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 165.8, (t, J = 41.1 Hz), 152.9, 131.3, 129.5, 120.8, (t, J = Hz), 82.4, 28.1; ( 19 F NMR, 377 MHz, CDCl 3): δ = (s); IR (neat): ν 3085, 3038, 3010, 2984, 2940, 2697, 2542, 1782, 1680, 1604, 1504, 1475, 1458, 1431, 1413, 1396, 1373, 1322, 1297, 1277, 1262, 1251, 1199, 1156, 1121, 1095, 1017, 977, 965, 927, 873, 842, 818, 769, 737, 715; MP: C; HRMS (ESI) for C 13H 14F 2Na5 [M+Na] + requires found (4-Benzoylphenoxy)(difluoro)acetic acid (3i) The title compound was prepared following general procedure D using ethyl (4- benzoylphenoxy)(difluoro)acetate (1.28 g, 4.0 mmol), 5 M aq. NaH (4 ml, 20.0 mmol) and Et 2 (20 ml); (1.05 mg, 95 % yield, white solid). ( 1 H NMR, 400 MHz, CDCl 3): δ = 8.59 (brs, 1H), 7.86 (d, J = 8.6 Hz, 2H), (d, J = 7.3 Hz, 2H), 7.64 (t, J = 7.3 Hz, 1H), 7.51 (t, J = 7.7 Hz, 2H), 7.35 (d, J = 8.6 Hz, 2H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 197.1, (t, J = 41.3 Hz), 153.0, 136.8, 134.9, 133.1, 132.2, 130.2, 128.5, 120.9, (t, J = Hz); ( 19 F NMR, 377 MHz, CDCl 3): δ = 77.0 (s); IR: ν 1774, 1645, 1598, 1503, 1448, 1279, 1168, 1016, 926, 851; HRMS (ESI) for C 15H 9F 2 4 [M-H] - requires found Data consistent with literature values. 2 (3-Cyanophenoxy)(difluoro)acetic acid (3j) The title compound was prepared following general procedure D using ethyl (3- cyanophenoxy)(difluoro)acetate (145 mg, 0.6 mmol), 5 M aq. NaH (0.6 ml, 3.0 mmol) and MeCN (30 ml); (125 mg, 98 % yield, white solid). ( 1 H NMR, 400 MHz, CDCl 3): δ = 9.94 (brs, 1H), (m, 4H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 40.5 Hz), 149.4, 130.9, 130.4, 126.7, 125.3, 117.2, (t, J = Hz), 113.5; ( 19 F NMR, 377 MHz, CDCl 3): δ = 77.2 (s); IR: ν 2242, 1770, 1582, 1483, 1433, 1239, 1160, 931; HRMS (ESI) for C 8H 4BrF 2 3 [M-H] - requires found ; Mp C. 20
21 Difluoro[4-(trifluoromethyl)phenoxy]acetic acid (3k) The title compound was prepared following general procedure D using ethyl difluoro[4- (trifluoromethyl)phenoxy]acetate (1.14 g, 4.0 mmol), 5 M aq. NaH (4 ml, 20.0 mmol) and MeCN (20 ml); (508 mg, 50 % yield, white solid). ( 1 H NMR, 400 MHz, CDCl 3): δ = 8.80 (brs, 1H), 7.67 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 8.6 Hz, 2H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = Hz), 151.7, (q, J = 33.1 Hz), (q, J = 3.7 Hz), (q, J = Hz), 121.7, (t, J =274.2 Hz); ( 19 F NMR, 377 MHz, CDCl 3): δ = 62.5 (s, 3F), 71.2 (s, 2F); IR: ν 1769, 1615, 1515, 1326, 1125, 1065, 1018, 846, 807; HRMS (EI/FI) for C 9H 5F 5 3 [M] + requires found ; Mp C. 2-(4-(1,3-Dioxoisoindolin-2-yl)phenoxy)-2,2-difluoroacetic acid (3l) and 2-((4-(Carboxydifluoromethoxy)phenyl)carbamoyl)benzoic acid (3l-2) Following general procedure D using ethyl [4-(1,3-dioxo-1,3-dihydro-2H-isoindol-2- yl)phenoxy](difluoro)acetate (60 mg, 0.17 mmol), aq. NaH (5.0 M; 170 µl, 0.83 mmol) and MeCN (830 μl) a mixture (40 mg) of two compounds 3l and 3l-2 was obtained in a ratio of 0.04:1.00 as determined by 1 H NMR as a white solid. ( 1 H NMR, 400 MHz, Acetone-d 6): δ = (m, 4H), (m, 2H), (m, 2H); ( 19 F NMR, 377 MHz, Acetone-d 6): δ = (s); HRMS (ESI) for C 16H 10F 2N 5 [M+H] + requires found ( 1 H NMR, 400 MHz, Acetone-d 6): δ = 7.97 (ddd, J = 7.6, 1.2, 0.5 Hz, 1H), (m, 2H), (m, 1H), (m, 2H), (m, 2H); ( 13 C NMR, 101 MHz, Acetone-d 6): δ = , , (t, J = 40.7 Hz), (t, J = 2.0 Hz), , , , , , , , , , (t, J = Hz); ( 19 F NMR, 377 MHz, Acetone-d 6): δ = (s); HRMS (ESI) for C 16H 12F 2N 6 [M+H] + requires found
22 2.5 Post-Fluorination Reactions N-(2-Fluoro-4-iodophenyl)methanesulfonamide The title compound was prepared following a procedure reported in the literature. 5 A cooled solution of 2-fluoro-4-iodoaniline (3.56 g, 15.0 mmol) in pyridine (20 ml) at 0 C was drop-wise treated with methanesulfonyl chloride (1.74 ml, 22.5 mmol). After being stirred for 3 h at room temperature, the mixture was diluted with water and extracted with ethyl acetate several times. The combined organic layers were washed with water and brine, dried over MgS 4, and concentrated in vacuo. The residue was purified by recrystalisation in DCM/n-pentane to afford the title compound (3.78 g, 80 % yield) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.42 (d, J = 8.3 Hz, 2H), 7.25 (t, J = 8.2 Hz, 1H), 6.70 (brs, 1H), 2.97 (s, 3H); ( 19 F NMR, 377 MHz, CDCl 3): δ = (t, J = 8.9 Hz). Data consistent with literature values. 5 N-(4-Cyano-2-fluorophenyl)methanesulfonamide The title compound was prepared following a procedure reported in the literature. 5 A mixture of N-(2- fluoro-4-iodophenyl)methanesulfonamide (2.52 g, 8 mmol), zinc cyanide (564 mg, 4.8 mmol), and tetrakis(triphenylphosphine) palladium (462 mg, 0.4 mmol) in dimethylformamide (18 ml) was heated at 80 C for 8 h. The mixture was diluted with water and extracted with ethyl acetate several times. The combined organic layers were washed with water and brine, dried over MgS 4, and concentrated in vacuo. The residue was purified by recrystalisation in DCM/n-pentane to afford the title compound (1.25 g, 73 % yield) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.66 (t, J = 8.2 Hz, 1H), (m, 2H), 6.91 (brs, 1H), 3.07 (s, 3H); ( 19 F NMR, 377 MHz, CDCl 3): δ = (t, J = 9.1 Hz). Data consistent with literature values. 5 (3-Fluoro-4-(methylsulfonamido)phenyl)methanaminium chloride The title compound was prepared following a procedure reported in the literature. 5 A stirred suspension of N-(4-cyano-2-fluorophenyl)methanesulfonamide (1.07 g, 5 mmol), 5% palladium on carbon (100 mg) and several drops of concentrated hydrochloric acid in MeH (25 ml) was hydrogenated under a balloon of hydrogen for 16 h. The reaction mixture was filtered and the filtrate was concentrated in vacuo to afford 3-fluoro-4-(methylsulfonylamino)benzyl amine salt as a white solid (1.02 g, 80 % yield). ( 1 H NMR, 400 MHz, CD 3D): δ = 7.49 (t, J = 8.3 Hz, 1H), 7.28 (dd, J1 = 2.0 Hz, J2 = 11.1 Hz, 1H), 7.21 (dd, J1 = 2.1 Hz, J2 = 8.3 Hz, 1H), 4.04 (s, 2H), 2.95 (s, 3H); ( 19 F NMR, 377 MHz, CD 3D): δ = (dd, J1 = 8.3 Hz, J2 = 11.1 Hz). Data consistent with literature values. 5 22
23 2-(4-(tert-Butyl)phenoxy)-2,2-difluoro-N-(3-fluoro-4-(methylsulfonamido)benzyl)acetamide (4) The title compound was prepared following a procedure reported in the literature. 6 To a solution of 4-tert-Butylphenoxy)(difluoro)acetic acid (48.8 mg, 0.2 mmol) in THF (2 ml) were added DMTMM (4- (4,6-dimethoxy-1,3,5- triazin-2-yl)-4-methylmorpholinium chloride) (86 mg, 0.3 mmol), NMM (23 μl, 0.2 mmol) and Et 3N (88 μl, 0.6 mmol). After being stirred for 4 h, the reaction mixture was added (3- fluoro-4-(methylsulfonamido)phenyl)methanaminium chloride (64 mg, 0.25 mmol). After additional 10 h, the reaction mixture was diluted with DCM, washed with water and brine, dried over MgS 4, and concentrated in vacuo. Purification of the residue by column chromatography (EtAc/n-pentane = 1:1) afforded the title compound (5.8 mg, 7 %) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.49 (t, J = 8.3 Hz, 1H), 7.31 (d, J = 8.8 Hz, 2H), (m, 4H), 6.82 (brs, 1H), 6.49 (brs, 1H), 4.46 (d, J = 6.1 Hz, 2H), 2.97 (s, 3H), 1.25 (s, 9H); ( 19 F NMR, 377 MHz, CDCl 3): δ = 77.0 (s, 2F), (t, J = 9.1 Hz, 1F). Data consistent with literature values. 6 2-Bromo-N,N-dimethylbenzenesulfonamide The title compound was prepared following a procedure reported in the literature. 7 To a solution of 2-bromobenzene-1-sulfonyl chloride (7.67 g, 30.0 mmol) in THF (20 ml) was slowly added dimethylamine (6.77 ml, 60.0 mmol) at 0 C. The reaction mixture was stirred at room temperature for 6 hours. The reaction was then poured into water and the mixture was extracted with DCM. The combined organic phases were washed with brine, dried over MgS 4 and concentrated in vacuo to provide the title compound (7.30 g, 92 % yield) as a beige solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 8.08 (dd, J 1 = 1.7 Hz, J 2 = 7.6 Hz, 1H), 7.75 (dd, J 1 = 1.2 Hz, J 2 = 7.8 Hz, 1H), 7.45 (td, J 1 = 1.2 Hz, J 2 = 7.6 Hz, 1H), 7.39 (td, J 1 = 1.7 Hz, J 2 = 7.6 Hz, 1H), 2.90 (s, 6H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 137.7, 135.7, 133.5, 132.3, 127.4, 120.3, Data consistent with literature values. 7 Di-tert-butyl (4-bromobenzene-1,2-diyl)biscarbamate To a solution of 4-bromobenzene-1,2-diamine (2.81 g, 15.0 mmol) in DCM (45 ml) was added di-tertbutyl dicarbonate (17.2 ml, 75.0 mmol) and 2M NaH (aq) (18.8 ml, 37.5 mmol). The reaction mixture was stirred at room temperature for 12 hours. The reaction mixture was extracted with DCM and the combined organic phases were washed with brine, dried over MgS 4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: 30% EtAc in n-hexane) to provide the title compound (4.88 g, 84 % yield) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.77 (s, 1H), 7.33 (s, 1H), 7.23 (dd, J 1 = 2.2 Hz, J 2 = 8.6 Hz, 1H), 6.74 (s, 1H), 6.56 (s, 1H), 1.52 (s, 9H), 1.52 (s, 9H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 153.7, 153.4, 146.8, 131.8, 127.9, 126.4, 125.7, 118.2, 81.3, 28.3; IR: ν 3300, 2980, 1700, 1594, 1515, 1413, 1393, 1368, 1238, 1155, 1051, 1024, 910, 855; HRMS (EI/FI) for C 16H 23N 2 4 [M] + requires found ; Mp C. Data consistent with literature values. 8 23
24 Di-tert-butyl (4-(2,2,5,5-tetramethylborolan-1-yl)-1,2-phenylene)dicarbamate To a mixture of bis(pinacolato)diboron (1.62 g, 6.38 mmol), Pd(dppf)Cl 2. CH 2Cl 2 (350 mg, 0.43 mmol) and potassium acetate (833 mg, 8.5 mmol) was added degassed DMF (10 ml) under N 2 atmosphere. To the reaction mixture was added di-tert-butyl (4-bromobenzene-1,2-diyl)biscarbamate (1.64 g, 4.25 mmol) and stirred at 80 C for 16 hours. The reaction mixture was concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: 10% EtAc in n-pentane) to provide the title compound (1.11 g, 60 % yield) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = (m, 2H), 7.60 (dd, J 1 = 1.0 Hz, J 2 = 8.1 Hz, 1H), 7.13 (s, 1H), 6.51 (s, 1H), 1.50 (s, 9H), 1.50 (s, 9H), 1.32 (s, 12H); ( 13 C NMR, 101 MHz, CDCl 3): δ = 154.2, 153.1, 135.0, 132.9, 131.6, 127.3, 121.7, 83.7, 80.9, 80.6, 28.2, 28.2, 24.8; IR: ν 3310, 2972, 1737, 1418, 1366, 1231, 1156, 1095, 855; HRMS (ESI) for C 22H 35 6N 2BNa [M+Na] + requires found ; Mp C. Data consistent with literature values. 9 Di-tert-butyl (2'-(N,N-dimethylsulfamoyl)-[1,1'-biphenyl]-3,4-diyl)dicarbamate To a solution of di-tert-butyl (4-(2,2,5,5-tetramethylborolan-1-yl)-1,2-phenylene)dicarbamate (109 mg, 0.25 mmol) and 2-bromo-N,N-dimethylbenzenesulfonamide (66.0 mg, 0.25 mmol) in DME (2.0 ml) and H 2 (0.5 ml) was added Na 2C 3 (126 mg, 1.5 mmol) and Pd(dppf)Cl 2. CH 2Cl 2 (20.4 mg, mmol). The reaction mixture was stirred at 100 C for 16 hours then concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: 30% Et 2 in n-pentane) to provide the title compound (84.4 mg, 69 % yield) as a yellow solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 8.03 (dd, J 1 = 1.2 Hz, J 2 = 7.8 Hz, 1H), (m, 4H), 7.24 (td, J 1 = 1.5 Hz, J 2 = 7.8 Hz, 1H), 7.11 (dd, J 1 = 2.0 Hz, J 2 = 8.3 Hz, 1H), 6.73 (brs, 2H), 2.32 (s, 6H), 1.46 (s, 9H), 1.43 (s, 9H); ( 13 C NMR, 126 MHz, CDCl 3): δ = 153.8, 153.7, 140.6, 137.2, 136.5, 132.9, 132.2, 130.4, 130.1, 129.3, 127.7, 126.6, 125.4, 123.0, 81.0, 36.0, 28.3, 28.3; IR: ν 3319, 2977, 2363, 1725, 1702, 1590, 1525, 1480, 1393, 1368, 1323; HRMS (EI/FI) for C 24H 33N 3 6S [M] + requires found ; Mp C. 3',4'-Diamino-N,N-dimethylbiphenyl-2-sulfonamide To a flask containing di-tert-butyl (2'-(N,N-dimethylsulfamoyl)-[1,1'-biphenyl]-3,4-diyl)dicarbamate (1.02 g, 2.08 mmol) was added 2M HCl in Et 2 (40 ml, 80 mmol) and 1.25 M HCl in MeH (16 ml, 20 mmol). The reaction mixture was stirred at room temperature for 4 hours then concentrated in vacuo. The residue was dissolved in DCM and the solution was washed with sat. NaHC 3(aq) and brine, dried over MgS 4 and concentrated in vacuo to provide the title compound (534 mg, 88 % yield) as a brown solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = 8.08 (dd, J 1 = 1.2 Hz, J 2 = 7.8 Hz, 1H), 7.50 (td, J 1 = 1.5 Hz, J 2 = 7.5 Hz, 1H), 7.41 (td, J 1 = 1.5 Hz, J 2 = 7.5 Hz, 1H), 7.28 (dd, J 1 = 1.2 Hz, J 2 = 7.8 Hz, 1H), 6.81 (t, J = 1.0 Hz, 1H), 6.87 (t, J = 1.2 Hz, 1H), 3.43 (brs, 4H), 2.38 (s, 6H); ( 13 C NMR, 126 MHz, CDCl 3): δ = 141.9, 137.4, 134.6, 24
25 133.7, 133.2, 132.0, 131.4, 130.3, 127.0, 121.4, 118.4, 115.7, 36.2; IR: ν 3417, 3349, 1627, 1587, 1517, 1465, 1314, 1276, 1153, 1071, 966, 816; HRMS (ESI) for C 14H 18 2N 3S [M+H] + requires found ; Mp C. 2-(2-{Difluoro[4-(trifluoromethyl)phenoxy]methyl}-1H-benzimidazol-5-yl)-N,Ndimethylbenzene sulfonamide (5) To a mixture of difluoro[4-(trifluoromethyl)phenoxy]acetic acid (218 mg, 0.85 mmol) and 3',4'- diamino-n,n-dimethylbiphenyl-2-sulfonamide (248 mg, 0.85 mmol) was added 6M HCl (aq) (2.0 ml). The reaction mixture was stirred at 110 C for 16 hours. The reaction mixture was concentrated in vacuo then purified by silica gel column chromatography (eluent: 30% EtAc in n-pentane) to provide the title compound (80.1 mg, 18 % yield) as a white solid. ( 1 H NMR, 400 MHz, CDCl 3): δ = (brs, 1H), 8.02 (dd, J 1 = 8.1 Hz, J 2 = 13.5 Hz, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.70 (d, J = 8.6 Hz, 1H), 7.59 (s, 1H), 7.57 (s, 1H), 7.52 (t, J = 7.3 Hz, 1H), 7.45 (td, J 1 = 1.2 Hz, J 2 = 7.6 Hz, 1H), (m, 2H), 7.28 (d, J = 7.6 Hz, 1H), (m, 1H), 2.32 (s, 3H), 2.24 (s, 3H); ( 13 C NMR, 126 MHz, CDCl 3) 152.0, (t, J = 39.1 Hz), 141.8, 141.4, 137.3, , 133.2, 132.3, (q, J = 33.1 Hz), (t, J = 6.2 Hz), (q, J = 3.6 Hz), 125.0, (q, J = Hz), 122.1, 121.2, 120.1, (t, J = Hz), 113.9, 110.8, 36.4, 36.2; ( 19 F NMR, 377 MHz, CDCl 3): δ = 62.3 (s, 3F), 65.9 (s, 2F); IR: ν 2974, 1742, 1449, 1370, 1322, 1220, 1162, 1125, 1066, 966; HRMS (ESI) for C 23H 18 3N 3F 5NaS [M+Na] + requires found ; Mp C. Data consistent with literature values tert-Butyl-4-(difluoromethoxy)benzene (6) To a solution of an appropriate (4-tert-butylphenoxy)(difluoro)acetic acid (488.4 mg, 2.0 mmol) in 1:1 acetone/water (8 ml) was added AgN 3 (339.8 mg, 2.0 mmol) and K 2S 2 8 (2.16 g, 8.0 mmol). The reaction mixture was stirred at 50 o C for 2 hours before diluting with water. The aqueous layer was extracted with DCM (3 x 10 ml), then the combined organic phases were washed with brine, dried over MgS 4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography (eluent: 1 % Et 2 in n-pentane) to provide the title compound (199.4 mg, 50 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.38 (dt, J 1 = 2.6 Hz, J 2 = 8.8 Hz, 2H), 7.06 (d, J = 8.8 Hz, 2H), 6.49 (t, J = 74.3 Hz, 1H), 1.33 (s, 9H); ( 13 C NMR, 101 MHz, CDCl 3): δ = (t, J = 2.8 Hz), 148.4, 126.7, 119.1, (t, J = Hz), 34.4, 31.4; ( 19 F NMR, 377 MHz, CDCl 3): δ = 80.4 (d, J = 74.6 Hz). Data consistent with literature values. 2 25
26 4-(4-(tert-Butyl)phenoxy)-4,4-difluorobutan-2-one (7) The title compound was isolated along with the synthesis of 1-tert-butyl-4-(difluoromethoxy)benzene (6). Purification by silica gel column chromatography (eluent: 1 % Et 2 in n-pentane) provided the title compound (16.3 mg, 3 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.37 (dt, J 1 = 2.6 Hz, J 2 = 8.8 Hz, 2H), 7.11 (d, J = 8.8 Hz, 2H), 3.26 (t, J = 10.6 Hz, 2H), 2.37 (s, 3H), 1.32 (s, 9H); ( 13 C NMR, 126 MHz, CDCl 3): δ = 200.1, 149.1, 148.0, 126.4, 120.7, 50.1, 34.5, 31.4, 30.3; ( 19 F NMR, 377 MHz, CDCl 3): δ = 65.9 (s, 2F); IR: 2964, 1722, 1510, 1412, 1365, 1335, 1265, 1202, 1171, 1121, 1017, 978; HRMS (ESI) for C 14H 18F 2Na 2 [M+Na] + requires found (tert-Butyl)-2-((4-(tert-butyl)phenoxy)difluoromethyl)-1-(difluoromethoxy)benzene (8) The title compound was isolated along with the synthesis of 1-tert-butyl-4-(difluoromethoxy)benzene (6). Purification by silica gel column chromatography (eluent: 1 % Et 2 in n-pentane) provided the title compound (14.0 mg, 4 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.79 (d, J = 2.5 Hz, 1H), 7.53 (dd, J 1 = 2.4 Hz, J 2 = 8.5 Hz, 1H), 7.39 (dt, J 1 = 2.5 Hz, J 2 = 8.8 Hz, 2H), (m, 3H), 6.50 (t, J = 74.7 Hz, 1H), 1.36 (s, 9H), 1.34 (s, 9H); ( 13 C NMR, 126 MHz, CDCl 3): δ = 200.1, 149.1, 148.0, 126.4, 120.7, 50.1, 34.5, 31.4, 30.3; ( 19 F NMR, 377 MHz, CDCl 3): δ = 65.7 (s, 2F), 79.9 (d, J = 74.6 Hz, 2F); IR: 2964, 1508, 1366, 1317, 1255, 1131, 1105, 1036; HRMS (EI/FI) for C 22H 26F 4 2 [M] + requires found (4-(tert-Butyl)phenoxy)propan-2-one (9) The title compound was isolated along with the synthesis of 1-tert-butyl-4-(difluoromethoxy)benzene (6). Purification by silica gel column chromatography (eluent: 1 % Et 2 in n-pentane) provided the title compound (15.8 mg, 4 % yield) as a colourless oil. ( 1 H NMR, 400 MHz, CDCl 3): δ = 7.41 (dt, J 1 = 2.6 Hz, J 2 = 8.8 Hz, 2H), 7.05 (dt, J 1 = 2.6 Hz, J 2 = 8.8 Hz, 2H), 3.70 (s, 2H), 2.37 (s, 3H), 1.32 (s, 9H); ( 13 C NMR, 126 MHz, CDCl 3): δ = 200.1, 149.1, 148.0, 126.4, 120.7, 50.1, 34.5, 31.4, 30.3; IR: 3402, 2962, 1742, 1613, 1515, 1207, 1179, 1145, 1110, 1016, 832; HRMS (EI/FI) for C 13H 18 2 [M] + requires found
27 3. ptimisation Table 3.1 ptimization for the synthesis of [ 18 F]2a from 1a (S1) entry solvent concentration (mm) additive (equiv) temp ( o C) t (min) [ 18 F]2a RCC (%) n 1 MeCN ± MeCN ± MeCN ± MeCN ± MeCN ± MeCN ± MeCN ± MeCN ± MeCN ± t-buh ± MeCN 100 AgN 3 (1.0) ± MeCN 100 NaI (1.0) ±
28 3.2 ptimization for the synthesis of [ 18 F]3a from 1a (S2) entry solvent conc. (mm) additive (equiv) temp ( o C) [ 18 F]2a RCC (%) [ 18 F]2a RCC (%) 1 MeCN 17 K 2C 3 (1.4) ± ± MeCN 17 K 2C 3 (2.8) ± 6 14 ± MeCN 17 K 2C 3 (1.4), K (2.4) 80 1 ± 1 82 ± MeCN 17 K 2C 3 (1.4), 18-crown-6 (2.4) ± ± MeCN 33 K 2C 3 (1.4), K (2.4) 80 < 1 73 ± a MeCN 17 K 2C 3 (1.4), K (2.4) 80 < 1 80 ± b MeCN 17 K 2C 3 (1.4), K (2.4) 80 < 1 80 ± a MeCN 17 K 2C 3 (1.4), K (1.4) 80 4 ± 3 70 ± MeCN 17 NEt 4HC 3 (1.4) ± 8 6 ± MeCN 17 NBu 4H (1.4) ± 0 1 ± MeCN 17 Cs 2C 3 (1.4) ± 1 36 ± MeCN 17 LiH (1.4) 80 9 ± 4 22 ± MeCN 17 CsH. H 2 (1.4) ± ± MeCN/H 2 (59:1) 17 Cs 2C 3 (1.4) ± 2 7 ± MeCN/H 2 (59:1) 17 LiH (1.4) ± 2 43 ± MeCN/H 2 (59:1) 17 CsH. H 2 (1.4) 80 5 ± 3 18 ± DMS 17 K 2C 3 (1.4), K (2.4) 80 < 1 2 ± DMF 17 K 2C 3 (1.4), K (2.4) 80 < 1 60 ± t-buh 17 K 2C 3 (1.4), K (2.4) 80 < 1 45 ± acetone 17 K 2C 3 (1.4), K (2.4) 50 < 1 81 ± acetone ± acetone 33 K 2C 3 (1.4), K (2.4) 50 < 1 74 ± acetone 50 K 2C 3 (1.4), K (2.4) 50 < 1 83 ± a acetone 17 K 2C 3 (1.4), K (2.4) 50 2 ± 1 79 ± a acetone 17 K 2C 3 (1.4), K (1.4) 50 4 ± 2 72 ± a acetone 17 K 2C 3 (1.4) ± 10 6 ± 1 2 a 10 min reaction; b 5 min reaction n 28
29 3.3 ptimization for the synthesis of [ 18 F]4 from [ 18 F]3a (S3) entry coupling reagent (mmol) additive (mmol) solvent [ 18 F]4 RCC (%) n 1 HATU (0.06) - DMF 8 ± HBTU (0.06) - DMS 2 ± DCC (0.06) HBt (0.04) DMF 6 ± 1 2 entry X coupling reagent (equiv) base (equiv) additive (equiv) [ 18 F]4 RCC (%) n DMTMM (0.06) NEt 3 (0.12) - 16 ± DMTMM (0.06) NEt 3 (0.12) NMM (0.04) 18 ± DMTMM (0.12) NEt 3 (0.24) - 12 ± DMTMM (0.12) NEt 3 (0.24) NMM (0.08) 10 ±
30 3.4 ptimization for the synthesis of 6 from 3a (S4) entry metal salt AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 Ag2C3 Ag2C3 Ag2C3 AgN3 AgN3 AgN3 4 equiv AgN3 4 equiv AgN3 Ag(pico)2 Ag(pico)2 AgN3 AgN3 Cu2 oxidant additive solvent temp 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv (NH4)2S28 4 equiv (NH4)2S28 4 equiv (NH4)2S28 4 equiv (NH4)2S28 4 equiv (NH4)2S28 6 (% 19 F) 3a (% 19 F) - 1:1 Acetone/H2 50 o C 53 % 1 % - 1:1 MeCN/H2 80 o C 6 % - - 1:1 THF/H2 80 o C 1 % 93 % - 1:1 dioxane/h2 100 o C 17 % 54 % - 1:1 DMF/H2 100 o C 9 % 36 % - 1:1 DMS/H2 100 o C 12 % 22 % K2C3 1:1 Acetone/H2 50 o C equiv K222 1:1 Acetone/H2 50 o C 9 % 84 % K2C3 2 equiv K222 1:1 Acetone/H2 50 o C 3 % 88 % - 1:1 Acetone/H2 50 o C 37 % 2 % - 1:1 MeCN/H2 80 o C 6 % - K2C3 1:1 Acetone/H2 50 o C :1 Acetone/H2 50 o C 41 % 1 % - 1:1 MeCN/H2 80 o C 7 % - K2C3 1:1 Acetone/H2 50 o C 39 % - K2C3 1:1 Acetone/H2 50 o C 30 % - K2C3 2 equiv K222 1:1 Acetone/H2 50 o C 3 % 97 % - - 1:1 Acetone/H2 50 o C - 45 % - - 1:1 MeCN/H2 80 o C equiv Selectfluor 4 equiv Selectfluor - 1:1 Acetone/H2 50 o C 33 % - - 1:1 MeCN/H2 80 o C 7 % Acetone 50 o C % 30
31 Cu2 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 AgN3 - - MeCN 80 o C % 2 equiv K2S28 K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28 4 equiv K2S28-1:1 Acetone/H2 50 o C 50 % 7 % - 1:1 Acetone/H2 50 o C 26 % 62 % HCl 1:1 Acetone/H2 50 o C % Hantzsch ester 1:1 Acetone/H2 50 o C 25 % 38 % 1,4 CHD 1:1 Acetone/H2 50 o C 1 % 99 % 1,3 CHD 1:1 Acetone/H2 50 o C 8 % 91 % CuI 1:1 Acetone/H2 50 o C 47 % 19 % - Acetone 50 o C % - 4:1 Acetone/H2 50 o C 5 % 119 % - 1:4 Acetone/H2 50 o C 38 % 2 % - 1:1 Acetone/H2 rt 4 % 78 % TEMP 1:1 Acetone/H2 50 o C 11 % 86 % 2 equiv TEMP 1:1 Acetone/H2 50 o C % - 1:1 Acetone/D2 50 o C 35 % 18 % - - 1:1 Acetone-d6/H2 1:1 Acetone-d6/D2 50 o C 50 o C 4 % + (5 %) a 3 % + (5 %) a 4 % 3 % 1:1 Acetone/H2 50 o C 4 % 96 % 1:1 Acetone/H2 50 o C 53 % 16 % 1:1 Acetone/H2 50 o C 35 % 11 % 1:1 Acetone/H2 50 o C 19 % 38 % H-Si(SiMe 3) 3 1:1 Acetone/H2 50 o C 1 % 66 % 1:1 THF/H2 50 o C Trace 95 % 1:1 MeCN/H2 80 o C 14 % Trace 1:1 Dioxane/H2 100 o C 20 % 80 % 31
32 53 AgN3 54 AgN3 55 AgN3 56 AgN3 57 AgN equiv K2S28 4 equiv K2S28 2 equiv Selectfluor 4 equiv Selectfluor 4 equiv Selectfluor 2 equiv XeF2 2 equiv XeF2 2 equiv XeF2 1:1 DMF/H2 100 o C 34 % 46 % 1:1 DMS/H2 100 o C 9 % 19 % - 1:1 Acetone/H2 50 o C 18 % 32 % - 1:1 Acetone/H2 50 o C - 1:1 MeCN/H2 80 o C - DCM rt 2 equiv TBAF DCM rt 33 % + (12 %) b 7 + (12 %) b 0 % + (25 %) b 0 % + (13 %) b 0 % 0 % 0 % 32 % 2 equiv HF.pyridine DCM rt 0 % 0 % a Yield of Aryl-CF 2D product; b Yield of Aryl-CF 3 product. 32
33 4. General Radiochemical Procedures 4.1 [ 18 F]Fluoride Preparation [ 18 F]Fluoride was produced by Erigal (Keele, UK) via the 18 (p,n) 18 F reaction and delivered as [ 18 F]fluoride in [ 18 ]water. Radiosynthesis and azeotropic drying was performed on a NanoTek automated microfluidic device (Advion). [ 18 F]Fluoride was separated from 18 -enriched-water using anion exchange cartridges (MP1, RTG, Tennessee, USA) and subsequently released with 550 μl of a solution of K 2.2.2/K 2C 3 (kryptofix (15 mg) and K 2C 3 (3 mg) in 1 ml of MeCN/H 2, 4:1) into the concentrator to form [ 18 F]KF/K The solution was dried with five cycles of azeotropic drying with acetonitrile (300 μl) and redissolved in anhydrous acetonitrile (1000 μl). [ 18 F]KF/K (ca. 30 MBq) was dispensed as aliquots of this stock solution into a v-vial containing the necessary reagents and a magnetic stirrer. 4.2 Analysis For all reactions, an aliquot was removed for analysis by radio-tlc and radio-hplc. Radio-TLC was carried out on Merck Kiesegel 60 F254 plates in EtAc/Hexane (1:1) or Methanol and analysed using a plastic scintillator/pmt detector. Radio-HPLC analysis was performed using a Dionex Ultimate 3000 dual channel HPLC system with a shared auto-sampler, parallel UV-detectors and LabLogic NaI/PMTradiodetectors with Flowram analogue output. A representative radio-hplc trace of the crude reaction mixture, with UV trace (254 nm) of authentic reference material overlaid, is shown below. HPLC Gradient Settings: Water + 0.1% TFA/ MeCN + 0.1% TFA, 1 ml/min, Waters Nova-Pak C18 Column, 4 μm, 3.9 x 150 mm: 0 1 min (5 % MeCN + 0.1% TFA) isocratic 1 11 min (5 % to 95 % MeCN + 0.1% TFA) linear increase min (95 % MeCN + 0.1% TFA) isocratic min (95 % to 5 % MeCN + 0.1% TFA) linear decrease min (5 % MeCN + 0.1% TFA) isocratic. Reported radiochemical conversions are decay corrected and calculated by radio-tlc taking into account the radiochemical purity as determined by radio-hplc. 4.3 Procedure for small scale [ 18 F]Fluorination-hydrolysis To a V-vial containing K 2C 3 (2 mg, mmol), K (9 mg, mmol) and a magnetic stirrer bar was added [ 18 F]KF/K in MeCN. A solution of appropriate starting material (0.01 mmol) in acetone (200 μl) was added via syringe. The sealed vial was heated at 50 C for 20 minutes, before being quenched with water (200 μl). An aliquot was removed for analysis by radio-tlc and radio-hplc for radiochemical yield and product identification. 33
![4.4 Procedure for determination of isolated RCY and SA of [ 18 F]3a [ 18 F]Fluoride was separated from 18 -enriched-water using an anion exchange cartridge (Waters Sep- Pak Accell Plus QMA Carbonate](/docs-images/92/108972095/images/34-0.jpg "Plus Light Cartridge, 46 mg Sorbent per Cartridge, 40 µm Particle Size, 50/pk (Part #186004540)) and subsequently released with 900 μl of a solution of K 2.")
34 4.4 Procedure for determination of isolated RCY and SA of [ 18 F]3a [ 18 F]Fluoride was separated from 18 -enriched-water using an anion exchange cartridge (Waters Sep- Pak Accell Plus QMA Carbonate Plus Light Cartridge, 46 mg Sorbent per Cartridge, 40 µm Particle Size, 50/pk (Part # )) and subsequently released with 900 μl of a solution of K 2.2.2/K 2C 3 (kryptofix (15 mg) and K 2C 3 (3 mg) in 1 ml MeCN/H 2, 4:1). The solution was dried with six cycles of azeotropic drying with acetonitrile (150 μl). To the v-vial containing the dried [ 18 F]KF/K residue was added a solution of ethyl bromo(4-tert-butylphenoxy)fluoroacetate in acetone (250 μl), which was then heated to 50 C for 20 minutes. After cooling to room temperature the reaction mixture was diluted into 8 ml of H 2 and loaded onto a Sep-Pak Light C18 cartridge (activated with 1 ml MeH followed by 10 ml of H 2), followed by washing with H 2 (2 x 1mL). 1 ml of air was then flushed through the cartridge, after which [ 18 F]3a was eluted with CH 3CN (3 x 500 l) to give a stock solution of [ 18 F]3a for post-fluorination studies. An aliquot of this solution was analysed by radio-hplc for to assess product identity and radiochemical purity. UV trace of authentic reference Crude radiohplc trace Two reactions were performed, starting from 5130 MBq and 2620 MBq [ 18 F]fluoride in [ 18 ]water; 1750 MBq and 971 MBq of 4-tert-butylphenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3a) were isolated respectively. RCY = 36 ± 2 % (n = 2). SA of [ 18 F]3a in the collected fraction was assessed by injection of an aliquot on an analytical Synergi 4µm Hydro-RP 80A, 150 x 4.6 mm with 45% MeCN + 0.1% TFA/55% H % TFA (isocratic 1 ml/min), monitoring with UV (220 nm) and radioactive traces. The SA was decay corrected to the end of synthesis. 34
![4.5 Procedure for determination of isolated RCY and SA of [ 18 F]3k [ 18 F]Fluoride was separated from 18 -enriched-water using an anion exchange cartridge (Waters Sep- Pak Accell Plus QMA Carbonate](/docs-images/92/108972095/images/35-0.jpg "Plus Light Cartridge, 46 mg Sorbent per Cartridge, 40 µm Particle Size, 50/pk (Part #186004540)) and subsequently released with 900 μl of a solution of K 2.")
35 4.5 Procedure for determination of isolated RCY and SA of [ 18 F]3k [ 18 F]Fluoride was separated from 18 -enriched-water using an anion exchange cartridge (Waters Sep- Pak Accell Plus QMA Carbonate Plus Light Cartridge, 46 mg Sorbent per Cartridge, 40 µm Particle Size, 50/pk (Part # )) and subsequently released with 900 μl of a solution of K 2.2.2/K 2C 3 (kryptofix (15 mg) and K 2C 3 (3 mg) in 1 ml MeCN/H 2, 4:1). The solution was dried with six cycles of azeotropic drying with acetonitrile (150 μl). To the v-vial containing the dried [ 18 F]KF/K residue was added a solution of ethyl bromo(4-tert-butylphenoxy)fluoroacetate in acetone (250 μl), which was then heated to 50 C for 20 minutes. After cooling to room temperature the reaction mixture was diluted into 8 ml of H 2 and loaded onto a Sep-Pak Light C18 cartridge (activated with 1 ml MeH followed by 10 ml of H 2), followed by washing with H 2 (2 x 1mL). 1 ml of air was then flushed through the cartridge, after which [ 18 F]3k was eluted with CH 3CN (3 x 500 l) to give a stock solution of [ 18 F]3k for post-fluorination studies. An aliquot of this solution was analysed by radio-hplc for to assess product identity and radiochemical purity. UV trace of authentic reference Crude radiohplc trace Two reactions were performed, starting from 5140 MBq and 2870 MBq [ 18 F]fluoride in [ 18 ]water; 1550 MBq and 900 MBq of 4-tert-butylphenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3k) were isolated respectively. RCY = 31 ± 1 % (n = 2). SA of [ 18 F]3k in the collected fraction was assessed by injection of an aliquot on an analytical Synergi 4µm Hydro-RP 80A, 150 x 4.6 mm with 35% MeCN + 0.1% TFA/65% H % TFA (isocratic 1 ml/min), monitoring with UV (220 nm) and radioactive traces. The SA was decay corrected to the end of synthesis. 35
([ 18")
![F]difluoro)acetate ([ 18 F]2a)](/docs-images/92/108972095/images/36-1.jpg "Ethyl (biphenyl-4-yloxy)([ 18")
![F]difluoro)acetate ([ 18 F]2b)](/docs-images/92/108972095/images/36-2.jpg "4-tert-Butylphenoxy)([ 18")
36 5. Radio-HPLC Traces Ethyl (4-tert-butylphenoxy)([ 18 F]difluoro)acetate ([ 18 F]2a) Ethyl (biphenyl-4-yloxy)([ 18 F]difluoro)acetate ([ 18 F]2b) 4-tert-Butylphenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3a) 36
![(Biphenyl-4-yloxy)([ 18 F]difluoro)acetic](/docs-images/92/108972095/images/37-0.jpg "acid ([ 18 F]3b) [ 18")
![F]Difluoro(phenoxy)acetic acid ([ 18](/docs-images/92/108972095/images/37-1.jpg "F]3c) (4-Bromophenoxy)([ 18")
37 (Biphenyl-4-yloxy)([ 18 F]difluoro)acetic acid ([ 18 F]3b) [ 18 F]Difluoro(phenoxy)acetic acid ([ 18 F]3c) (4-Bromophenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3d) 37
![(3-Bromophenoxy)([ 18 F]difluoro)acetic acid](/docs-images/92/108972095/images/38-0.jpg "([ 18 F]3e) (2-Bromophenoxy)( [ 18")
![F]difluoro)acetic acid ([ 18 F]3f) 2,2-[ 18](/docs-images/92/108972095/images/38-1.jpg "F]Difluoro-2-(2-fluorophenoxy)acetic acid ([")
38 (3-Bromophenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3e) (2-Bromophenoxy)( [ 18 F]difluoro)acetic acid ([ 18 F]3f) 2,2-[ 18 F]Difluoro-2-(2-fluorophenoxy)acetic acid ([ 18 F]3g) 38
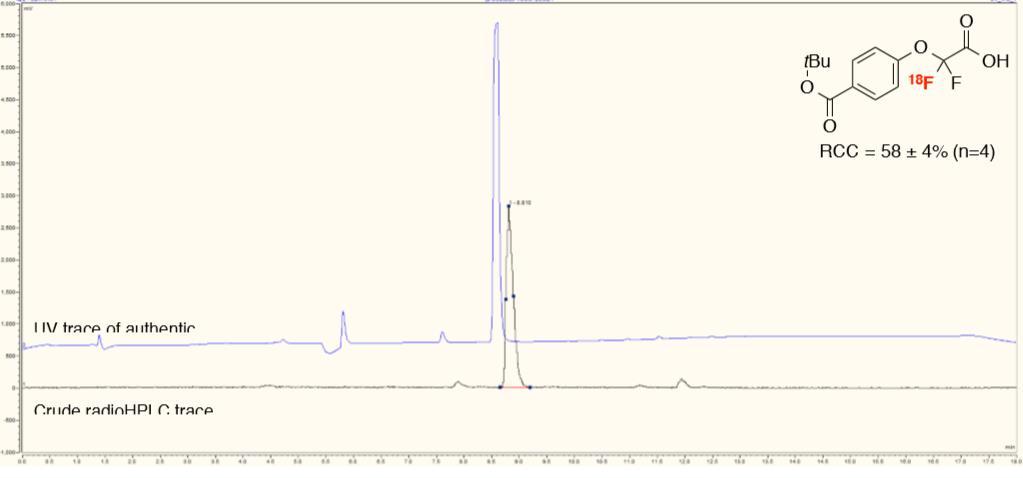
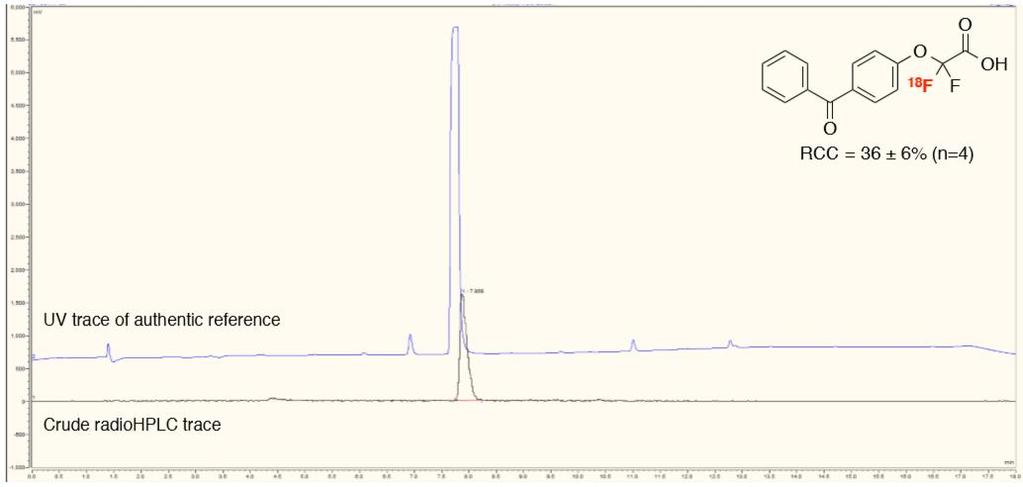
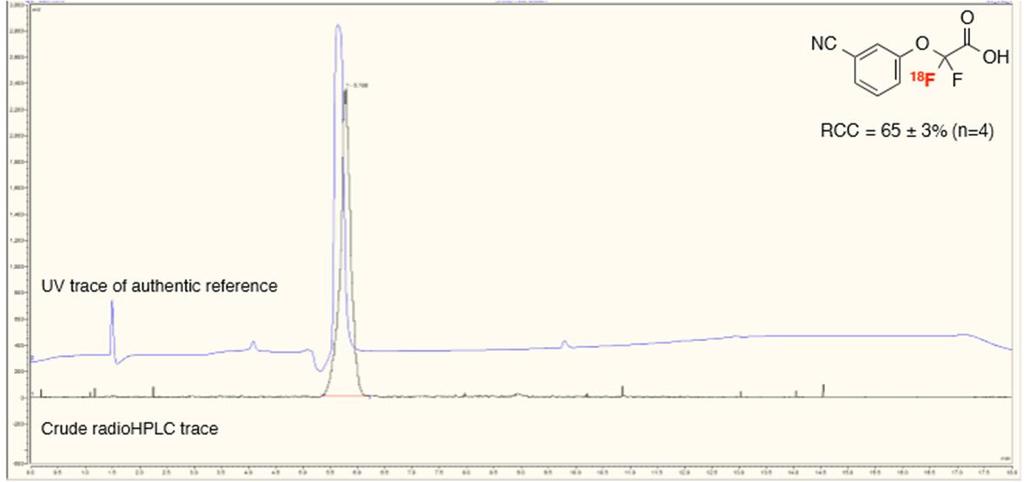
39 2-(4-(tert-Butoxycarbonyl)phenoxy)-2,2-[ 18 F]difluoroacetic acid ([ 18 F]3h) (4-Benzoylphenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3i) (3-Cyanophenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3j) 39
![[ 18 F]Difluoro[4-(trifluoromethyl)phenoxy]acetic acid ([ 18 F]3k)](/docs-images/92/108972095/images/40-1.jpg "2-(4-(1,3-Dioxoisoindolin-2-yl)phenoxy)-2,2-[ 18 F]difluoroacetic acid ([ 18 F]3l) and 2-((4-(Carboxy[ 18")
40 [ 18 F]Difluoro[4-(trifluoromethyl)phenoxy]acetic acid ([ 18 F]3k) 2-(4-(1,3-Dioxoisoindolin-2-yl)phenoxy)-2,2-[ 18 F]difluoroacetic acid ([ 18 F]3l) and 2-((4-(Carboxy[ 18 F]difluoromethoxy)phenyl)carbamoyl)benzoic acid 40
phenoxy)-2,2-[ 18 F]difluoro-N-(3-fluoro-4-(methyl sulfonamido)benzyl)acetamide ([ 18 F]4) To a V-vial containing a magnetic stirrer bar and")
41 6. Post-Fluorination Functionalization 6.1 Radiosynthesis of 2-(4-(tert-Butyl)phenoxy)-2,2-[ 18 F]difluoro-N-(3-fluoro-4-(methyl sulfonamido)benzyl)acetamide ([ 18 F]4) To a V-vial containing a magnetic stirrer bar and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4- methylmorpholinium chloride (DMTMM) (16.6 mg, 0.06 mmol) and (3-fluoro-4- (methylsulfonamido)phenyl)methanaminium chloride (10 mg, 0.04 mmol) was added 4-tertbutylphenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3a) in MeCN (~30 MBq). A solution of NEt 3 (17 μl, 0.12 mmol) in THF (300 μl) was added, after which the sealed vial was allowed to stir for 20 minutes at room temperature. After the reaction, an aliquot was removed for analysis by radio-tlc and HPLC to determine radiochemical conversion and product identity. Analysis was performed using Gradient A with a Phenomenex Synergi Hydro RP 80 Å column (4 μm, 4.6 x 150 mm) at a flow rate 1 ml/min. 2-(4-(tert-Butyl)phenoxy)-2,2-[ 18 F]difluoro-N-(3-fluoro-4-(methylsulfonamido)benzyl)acetamide ([ 18 F]4) UV trace of authentic reference Crude radiohplc trace 41
![6.2 Radiosynthesis of 2-(2-{[ 18 F]difluoro[4-(trifluoromethyl)phenoxy]methyl}-1Hbenzimidazol-5-yl)-N,N-dimethylbenzene sulfonamide ([ 18 F]5) To a V-vial containing a magnetic stirrer bar was added](/docs-images/92/108972095/images/42-0.jpg "[ 18 F]difluoro[4-(trifluoromethyl)phenoxy]acetic acid ([ 18 F]3k) in MeCN (~30 MBq). The solvent was then removed at 50 C under a stream of nitrogen for approximately 5 minutes.")
42 6.2 Radiosynthesis of 2-(2-{[ 18 F]difluoro[4-(trifluoromethyl)phenoxy]methyl}-1Hbenzimidazol-5-yl)-N,N-dimethylbenzene sulfonamide ([ 18 F]5) To a V-vial containing a magnetic stirrer bar was added [ 18 F]difluoro[4-(trifluoromethyl)phenoxy]acetic acid ([ 18 F]3k) in MeCN (~30 MBq). The solvent was then removed at 50 C under a stream of nitrogen for approximately 5 minutes. A solution of 3',4'-diamino-N,N-dimethylbiphenyl-2-sulfonamide in 6 M HCl (200 μl) was added, then the sealed vial was allowed to stir for 20 minutes at 110 o C. After the reaction, an aliquot was removed for analysis by radio-tlc and HPLC to determine radiochemical conversion and product identity. Analysis was performed using Gradient A with a Phenomenex Synergi Hydro RP 80 Å column (4 μm, 4.6 x 150 mm) at a flow rate 1 ml/min. 2-(2-{[ 18 F]Difluoro[4-(trifluoromethyl)phenoxy]methyl}-1H-benzimidazol-5-yl)-N,Ndimethylbenzene sulfonamide ([ 18 F]5) UV trace of authentic reference Crude radiohplc trace 42
![6.3 Radiosynthesis and isolation of 1-tert-butyl-4-([ 18 F]difluoromethoxy)benzene ([ 18 F]6) To a V-vial containing a magnetic stirrer bar was added 4-(tert-butylphenoxy)([ 18 F]difluoro)acetic acid](/docs-images/92/108972095/images/43-0.jpg "([ 18 F]3a) in MeCN (~30 MBq). The solvent was then removed at 50 C under a stream of nitrogen for approximately 5 minutes. Then acetone (200 μl) was added followed by a solution of AgN 3 (0.2 mg, 0.")
43 6.3 Radiosynthesis and isolation of 1-tert-butyl-4-([ 18 F]difluoromethoxy)benzene ([ 18 F]6) To a V-vial containing a magnetic stirrer bar was added 4-(tert-butylphenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3a) in MeCN (~30 MBq). The solvent was then removed at 50 C under a stream of nitrogen for approximately 5 minutes. Then acetone (200 μl) was added followed by a solution of AgN 3 (0.2 mg, 0.01 mmol) in H 2 (100 μl) and a solution of K 2S 2 8 (11 mg, 0.04 mmol) in H 2 (100 μl). The sealed vial was allowed to stir for 20 minutes at 50 C. After the reaction, the mixture was injected onto a reverse phase HPLC column (Phenomenex Luna C18(2) 100Å, 250 x 10mm). Flow rate 4 ml/min (70 % MeCN) [ 18 F]1-tert-butyl-4-(difluoromethoxy)benzene ([ 18 F]6) was eluted at 16.5 min. Two reactions were performed, starting from 1756 MBq and 836 MBq of 4-tertbutylphenoxy)([ 18 F]difluoro)acetic acid ([ 18 F]3a) (after solvent removal); 408 MBq and 179 MBq of 1- tert-butyl-4-([ 18 F]difluoromethoxy)benzene ([ 18 F]6) were isolated respectively. RCY = 22 ± 1 % (n = 2). 1-tert-Butyl-4-([ 18 F]difluoromethoxy)benzene ([ 18 F]6): Eluent system = 70 % MeCN isocratic UV trace of authentic reference Crude radiohplc trace SA of [ 18 F]6 in the collected fraction was assessed by injection of an aliquot on an analytical Synergi 4µm Hydro-RP 80A, 150 x 4.6 mm with 45% MeCN + 0.1% TFA/55% H % TFA (isocratic 1 ml/min), monitoring with UV (220 nm) and radioactive traces. The SA was decay corrected to the end of synthesis. 43
Direct Transformation of Ethylarenes into Primary Aromatic Amides with N-Bromosuccinimide and I 2 -aq NH 3
 Supporting Information Direct Transformation of Ethylarenes into Primary Aromatic Amides with N-Bromosuccinimide and I 2 -aq NH 3 Shohei Shimokawa, Yuhsuke Kawagoe, Katsuhiko Moriyama, Hideo Togo* Graduate
Supporting Information Direct Transformation of Ethylarenes into Primary Aromatic Amides with N-Bromosuccinimide and I 2 -aq NH 3 Shohei Shimokawa, Yuhsuke Kawagoe, Katsuhiko Moriyama, Hideo Togo* Graduate
A facile and general route to 3-((trifluoromethyl)thio)benzofurans and 3-((trifluoromethyl)thio)benzothiophenes
thio)benzofurans and 3-((trifluoromethyl)thio)benzothiophenes") Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2014 A facile and general route to 3-((trifluoromethyl)thio)benzofurans and 3-((trifluoromethyl)thio)benzothiophenes
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2014 A facile and general route to 3-((trifluoromethyl)thio)benzofurans and 3-((trifluoromethyl)thio)benzothiophenes
Copper-catalyzed formal O-H insertion reaction of α-diazo-1,3-dicarb- onyl compounds to carboxylic acids with the assistance of isocyanide
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2014 Copper-catalyzed formal O-H insertion reaction of α-diazo-1,3-dicarb- onyl compounds to carboxylic
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2014 Copper-catalyzed formal O-H insertion reaction of α-diazo-1,3-dicarb- onyl compounds to carboxylic
Supporting Information
 Supporting Information for Lewis acid-catalyzed redox-neutral amination of 2-(3-pyrroline-1-yl)benzaldehydes via intramolecular [1,5]-hydride shift/isomerization reaction Chun-Huan Jiang, Xiantao Lei,
Supporting Information for Lewis acid-catalyzed redox-neutral amination of 2-(3-pyrroline-1-yl)benzaldehydes via intramolecular [1,5]-hydride shift/isomerization reaction Chun-Huan Jiang, Xiantao Lei,
Supporting Information
 Supporting Information Copper/Silver Cocatalyzed Oxidative Coupling of Vinylarenes with ICH 2 CF 3 or ICH 2 CHF 2 Leading to β-cf 3 /CHF 2 -Substituted Ketones Niannian Yi, Hao Zhang, Chonghui Xu, Wei
Supporting Information Copper/Silver Cocatalyzed Oxidative Coupling of Vinylarenes with ICH 2 CF 3 or ICH 2 CHF 2 Leading to β-cf 3 /CHF 2 -Substituted Ketones Niannian Yi, Hao Zhang, Chonghui Xu, Wei
Highly enantioselective cascade synthesis of spiropyrazolones. Supporting Information. NMR spectra and HPLC traces
 Highly enantioselective cascade synthesis of spiropyrazolones Alex Zea a, Andrea-Nekane R. Alba a, Andrea Mazzanti b, Albert Moyano a and Ramon Rios a,c * Supporting Information NMR spectra and HPLC traces
Highly enantioselective cascade synthesis of spiropyrazolones Alex Zea a, Andrea-Nekane R. Alba a, Andrea Mazzanti b, Albert Moyano a and Ramon Rios a,c * Supporting Information NMR spectra and HPLC traces
Vilsmeier Haack reagent-promoted formyloxylation of α-chloro-narylacetamides
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 205 Vilsmeier aack reagent-promoted formyloxylation of α-chloro-arylacetamides by formamide Jiann-Jyh
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 205 Vilsmeier aack reagent-promoted formyloxylation of α-chloro-arylacetamides by formamide Jiann-Jyh
Supporting Information
 Supporting Information for AgOTf-catalyzed one-pot reactions of 2-alkynylbenzaldoximes with α,β-unsaturated carbonyl compounds Qiuping Ding 1, Dan Wang 1, Puying Luo* 2, Meiling Liu 1, Shouzhi Pu* 3 and
Supporting Information for AgOTf-catalyzed one-pot reactions of 2-alkynylbenzaldoximes with α,β-unsaturated carbonyl compounds Qiuping Ding 1, Dan Wang 1, Puying Luo* 2, Meiling Liu 1, Shouzhi Pu* 3 and
Site-Selective Suzuki-Miyaura Cross-Coupling Reactions of 2,3,4,5-Tetrabromofuran
 1 Site-Selective Suzuki-Miyaura Cross-Coupling Reactions of 2,3,4,5-Tetrabromofuran Munawar Hussain, a Rasheed Ahmad Khera, a Nguyen Thai Hung, a Peter Langer* a,b a Institut für Chemie, Universität Rostock,
1 Site-Selective Suzuki-Miyaura Cross-Coupling Reactions of 2,3,4,5-Tetrabromofuran Munawar Hussain, a Rasheed Ahmad Khera, a Nguyen Thai Hung, a Peter Langer* a,b a Institut für Chemie, Universität Rostock,
Eco-friendly synthesis of diverse and valuable 2-pyridones by catalyst- and solvent-free thermal multicomponent domino reaction
 Electronic Supplementary Material (ESI) for Green Chemistry. This journal is The Royal Society of Chemistry 2015 SUPPRTIG IFRMATI Eco-friendly synthesis of diverse and valuable 2-pyridones by catalyst-
Electronic Supplementary Material (ESI) for Green Chemistry. This journal is The Royal Society of Chemistry 2015 SUPPRTIG IFRMATI Eco-friendly synthesis of diverse and valuable 2-pyridones by catalyst-
Fluorinative Ring-opening of Cyclopropanes by Hypervalent Iodine Reagents. An Efficient Method for 1,3- Oxyfluorination and 1,3-Difluorination
 Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2016 Supporting Information Fluorinative Ring-opening of Cyclopropanes by Hypervalent Iodine
Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2016 Supporting Information Fluorinative Ring-opening of Cyclopropanes by Hypervalent Iodine
Direct Palladium-Catalyzed Arylations of Aryl Bromides. with 2/9-Substituted Pyrimido[5,4-b]indolizines
![Direct Palladium-Catalyzed Arylations of Aryl Bromides. with 2/9-Substituted Pyrimido[5,4-b]indolizines](/thumbs/87/95187440.jpg "Direct Palladium-Catalyzed Arylations of Aryl Bromides. with 2/9-Substituted Pyrimido[5,4-b]indolizines") Direct Palladium-Catalyzed Arylations of Aryl Bromides with 2/9-Substituted Pyrimido[5,4-b]indolizines Min Jiang, Ting Li, Linghua Meng, Chunhao Yang,* Yuyuan Xie*, and Jian Ding State Key Laboratory of
Direct Palladium-Catalyzed Arylations of Aryl Bromides with 2/9-Substituted Pyrimido[5,4-b]indolizines Min Jiang, Ting Li, Linghua Meng, Chunhao Yang,* Yuyuan Xie*, and Jian Ding State Key Laboratory of
Supporting information
 Electronic upplementary Material (EI) for New Journal of Chemistry. This journal is The Royal ociety of Chemistry and the Centre National de la Recherche cientifique 7 upporting information Lipase catalyzed,-addition
Electronic upplementary Material (EI) for New Journal of Chemistry. This journal is The Royal ociety of Chemistry and the Centre National de la Recherche cientifique 7 upporting information Lipase catalyzed,-addition
Supporting Information
 Supporting Information Ceric Ammonium Nitrate (CAN) catalyzed efficient one-pot three component aza-diels-alder reactions for a facile synthesis of tetrahydropyranoquinoline derivatives Ravinder Goud Puligoundla
Supporting Information Ceric Ammonium Nitrate (CAN) catalyzed efficient one-pot three component aza-diels-alder reactions for a facile synthesis of tetrahydropyranoquinoline derivatives Ravinder Goud Puligoundla
Supplementary information
 Electronic Supplementary Material (ESI) for MedChemComm. This journal is The Royal Society of Chemistry 2015 Supplementary information Synthesis of carboxyimidamide-substituted benzo[c][1,2,5]oxadiazoles
Electronic Supplementary Material (ESI) for MedChemComm. This journal is The Royal Society of Chemistry 2015 Supplementary information Synthesis of carboxyimidamide-substituted benzo[c][1,2,5]oxadiazoles
Regioselectivity in the Stille coupling reactions of 3,5- dibromo-2-pyrone.
 Regioselectivity in the Stille coupling reactions of 3,5- dibromo-2-pyrone. Won-Suk Kim, Hyung-Jin Kim and Cheon-Gyu Cho Department of Chemistry, Hanyang University, Seoul 133-791, Korea Experimental Section
Regioselectivity in the Stille coupling reactions of 3,5- dibromo-2-pyrone. Won-Suk Kim, Hyung-Jin Kim and Cheon-Gyu Cho Department of Chemistry, Hanyang University, Seoul 133-791, Korea Experimental Section
Electronic Supplementary Information
 Electronic Supplementary Information Unprecedented Carbon-Carbon Bond Cleavage in Nucleophilic Aziridine Ring Opening Reaction, Efficient Ring Transformation of Aziridines to Imidazolidin-4-ones Jin-Yuan
Electronic Supplementary Information Unprecedented Carbon-Carbon Bond Cleavage in Nucleophilic Aziridine Ring Opening Reaction, Efficient Ring Transformation of Aziridines to Imidazolidin-4-ones Jin-Yuan
Supporting Information
 Supporting Information Wiley-VC 007 9 Weinheim, Germany ew ear Infrared Dyes and Fluorophores Based on Diketopyrrolopyrroles Dipl.-Chem. Georg M. Fischer, Dipl.-Chem. Andreas P. Ehlers, Prof. Dr. Andreas
Supporting Information Wiley-VC 007 9 Weinheim, Germany ew ear Infrared Dyes and Fluorophores Based on Diketopyrrolopyrroles Dipl.-Chem. Georg M. Fischer, Dipl.-Chem. Andreas P. Ehlers, Prof. Dr. Andreas
Copper-Catalyzed Oxidative Dehydrogenative N-N Bond. Formation for the Synthesis of N,N -Diarylindazol-3-ones
 Electronic Supplementary Material (ESI) for Organic Chemistry Frontiers. This journal is the Partner Organisations 2016 Supporting information Copper-Catalyzed Oxidative Dehydrogenative - Bond Formation
Electronic Supplementary Material (ESI) for Organic Chemistry Frontiers. This journal is the Partner Organisations 2016 Supporting information Copper-Catalyzed Oxidative Dehydrogenative - Bond Formation
Supporting Information. Experimental section
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 Supporting Information Experimental section General. Anhydrous solvents were transferred by
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 Supporting Information Experimental section General. Anhydrous solvents were transferred by
Enantioselective Organocatalytic Michael Addition of Isorhodanines. to α, β-unsaturated Aldehydes
 Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2016 Enantioselective Organocatalytic Michael Addition of Isorhodanines to α,
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2016 Enantioselective Organocatalytic Michael Addition of Isorhodanines to α,
Supporting information
 Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2014 Supporting information Copper-catalysed intramolecular O-arylation: a simple
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2014 Supporting information Copper-catalysed intramolecular O-arylation: a simple
Supporting Information. Copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006
 Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006 1 A Facile Way to Synthesize 2H-Chromenes: Reconsideration of the Reaction Mechanism between Salicylic Aldehyde and
Supporting Information Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006 1 A Facile Way to Synthesize 2H-Chromenes: Reconsideration of the Reaction Mechanism between Salicylic Aldehyde and
Divergent synthesis of various iminocyclitols from D-ribose
 Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 205 Divergent synthesis of various iminocyclitols from D-ribose Ramu Petakamsetty,
Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 205 Divergent synthesis of various iminocyclitols from D-ribose Ramu Petakamsetty,
Supporting Information One-Pot Approach to Chiral Chromenes via Enantioselective Organocatalytic Domino Oxa-Michael-Aldol Reaction
 Supporting Information ne-pot Approach to Chiral Chromenes via Enantioselective rganocatalytic Domino xa-michael-aldol Reaction Hao Li, Jian Wang, Timiyin E-Nunu, Liansuo Zu, Wei Jiang, Shaohua Wei, *
Supporting Information ne-pot Approach to Chiral Chromenes via Enantioselective rganocatalytic Domino xa-michael-aldol Reaction Hao Li, Jian Wang, Timiyin E-Nunu, Liansuo Zu, Wei Jiang, Shaohua Wei, *
Supporting Information. Asymmetric Binary-acid Catalysis with Chiral. Phosphoric Acid and MgF 2 : Catalytic
 Supporting Information Asymmetric Binary-acid Catalysis with Chiral Phosphoric Acid and MgF 2 : Catalytic Enantioselective Friedel-Crafts Reactions of β,γ- Unsaturated-α-Ketoesters Jian Lv, Xin Li, Long
Supporting Information Asymmetric Binary-acid Catalysis with Chiral Phosphoric Acid and MgF 2 : Catalytic Enantioselective Friedel-Crafts Reactions of β,γ- Unsaturated-α-Ketoesters Jian Lv, Xin Li, Long
Aminofluorination of Fluorinated Alkenes
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2018 Synthesis of ɑ CF 3 and ɑ CF 2 H Amines via Aminofluorination of Fluorinated Alkenes Ling Yang,
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2018 Synthesis of ɑ CF 3 and ɑ CF 2 H Amines via Aminofluorination of Fluorinated Alkenes Ling Yang,
The Free Internet Journal for Organic Chemistry
 The Free Internet Journal for Organic Chemistry Paper Archive for Organic Chemistry Arkivoc 2018, part iii, S1-S6 Synthesis of dihydropyranones and dihydropyrano[2,3- d][1,3]dioxine-diones by cyclization
The Free Internet Journal for Organic Chemistry Paper Archive for Organic Chemistry Arkivoc 2018, part iii, S1-S6 Synthesis of dihydropyranones and dihydropyrano[2,3- d][1,3]dioxine-diones by cyclization
Supporting Information
 Supporting Information Lewis acid catalyzed ring-opening reactions of methylenecyclopropanes with diphenylphosphine oxide in the presence of sulfur or selenium Min Shi,* Min Jiang and Le-Ping Liu State
Supporting Information Lewis acid catalyzed ring-opening reactions of methylenecyclopropanes with diphenylphosphine oxide in the presence of sulfur or selenium Min Shi,* Min Jiang and Le-Ping Liu State
Electronic Supplementary Information (ESI)
") Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry Electronic Supplementary Information (ESI) For Iron-Catalysed xidative Amidation of Alcohols with Amines Silvia Gaspa, a Andrea
Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry Electronic Supplementary Information (ESI) For Iron-Catalysed xidative Amidation of Alcohols with Amines Silvia Gaspa, a Andrea
Room Temperature Highly Diastereoselective Zn-Mediated. Allylation of Chiral N-tert-Butanesulfinyl Imines: Remarkable Reaction Condition Controlled
 Supporting Information for: Room Temperature Highly Diastereoselective Zn-Mediated Allylation of Chiral N-tert-Butanesulfinyl Imines: Remarkable Reaction Condition Controlled Stereoselectivity Reversal
Supporting Information for: Room Temperature Highly Diastereoselective Zn-Mediated Allylation of Chiral N-tert-Butanesulfinyl Imines: Remarkable Reaction Condition Controlled Stereoselectivity Reversal
Supporting Information. Synthesis and biological evaluation of 2,3-Bis(het)aryl-4-azaindoles Derivatives as protein kinases inhibitors
aryl-4-azaindoles Derivatives as protein kinases inhibitors") Supporting Information Synthesis and biological evaluation of 2,3-Bis(het)aryl-4-azaindoles Derivatives as protein kinases inhibitors Frédéric Pin, a Frédéric Buron, a Fabienne Saab, a Lionel Colliandre,
Supporting Information Synthesis and biological evaluation of 2,3-Bis(het)aryl-4-azaindoles Derivatives as protein kinases inhibitors Frédéric Pin, a Frédéric Buron, a Fabienne Saab, a Lionel Colliandre,
Facile construction of the functionalized 4H-chromene via tandem. benzylation and cyclization. Jinmin Fan and Zhiyong Wang*
 Facile construction of the functionalized 4H-chromene via tandem benzylation and cyclization Jinmin Fan and Zhiyong Wang* Hefei National Laboratory for Physical Science at Microscale, Joint- Lab of Green
Facile construction of the functionalized 4H-chromene via tandem benzylation and cyclization Jinmin Fan and Zhiyong Wang* Hefei National Laboratory for Physical Science at Microscale, Joint- Lab of Green
Supporting Information for
 Supporting Information for An atom-economic route to densely functionalized thiophenes via base-catalyzed rearrangement of 5-propargyl-2H-thiopyran-4(3H)-ones Chunlin Tang a, Jian Qin b, Xingqi Li *a a
Supporting Information for An atom-economic route to densely functionalized thiophenes via base-catalyzed rearrangement of 5-propargyl-2H-thiopyran-4(3H)-ones Chunlin Tang a, Jian Qin b, Xingqi Li *a a
First DMAP-mediated direct conversion of Morita Baylis. Hillman alcohols into γ-ketoallylphosphonates: Synthesis of
 Supporting Information File 1 for First DMAP-mediated direct conversion of Morita Baylis Hillman alcohols into γ-ketoallylphosphonates: Synthesis of γ-aminoallylphosphonates Marwa Ayadi 1,2, Haitham Elleuch
Supporting Information File 1 for First DMAP-mediated direct conversion of Morita Baylis Hillman alcohols into γ-ketoallylphosphonates: Synthesis of γ-aminoallylphosphonates Marwa Ayadi 1,2, Haitham Elleuch
Supporting Information. Table of Contents. II. Experimental procedures. II. Copies of 1H and 13C NMR spectra for all compounds
 Electronic upplementary Material (EI) for rganic & Biomolecular Chemistry. This journal is The Royal ociety of Chemistry 2017 Laboratoire de Méthodologie et ynthèse de Produit aturels. Université du Québec
Electronic upplementary Material (EI) for rganic & Biomolecular Chemistry. This journal is The Royal ociety of Chemistry 2017 Laboratoire de Méthodologie et ynthèse de Produit aturels. Université du Québec
Supporting Information
 Supporting Information An Approach to 3,6-Disubstituted 2,5-Dioxybenzoquinones via Two Sequential Suzuki Couplings. Three-step Synthesis of Leucomelone Xianwen Gan, Wei Jiang, Wei Wang,,,* Lihong Hu,,*
Supporting Information An Approach to 3,6-Disubstituted 2,5-Dioxybenzoquinones via Two Sequential Suzuki Couplings. Three-step Synthesis of Leucomelone Xianwen Gan, Wei Jiang, Wei Wang,,,* Lihong Hu,,*
Copper-mediated radical cross-coupling reaction of 2,2-dichloro-1,1,1-trifluoroethane (HCFC-123) with phenols or thiophenols. Support Information
 with phenols or thiophenols. Support Information") Copper-mediated radical cross-coupling reaction of 2,2-dichloro-1,1,1-trifluoroethane (HCFC-123) with phenols or thiophenols Dr. Xiao un Tang and Prof. Qing un Chen* Key Laboratory of Organofluorine Chemistry,
Copper-mediated radical cross-coupling reaction of 2,2-dichloro-1,1,1-trifluoroethane (HCFC-123) with phenols or thiophenols Dr. Xiao un Tang and Prof. Qing un Chen* Key Laboratory of Organofluorine Chemistry,
Supporting Information for Iron-catalyzed decarboxylative alkenylation of cycloalkanes with arylvinylic carboxylic acids via a radical process
 Supporting Information for Iron-catalyzed decarboxylative alkenylation of cycloalkanes with arylvinylic carboxylic acids via a radical process Jincan Zhao 1, Hong Fang 1, Jianlin Han* 1,2 and Yi Pan* 1
Supporting Information for Iron-catalyzed decarboxylative alkenylation of cycloalkanes with arylvinylic carboxylic acids via a radical process Jincan Zhao 1, Hong Fang 1, Jianlin Han* 1,2 and Yi Pan* 1
Supporting Information
 Supporting Information Montmorillonite KSF-Catalyzed One-pot, Three-component, Aza-Diels- Alder Reactions of Methylenecyclopropanes With Arylaldehydes and Aromatic Amines Li-Xiong Shao and Min Shi* General
Supporting Information Montmorillonite KSF-Catalyzed One-pot, Three-component, Aza-Diels- Alder Reactions of Methylenecyclopropanes With Arylaldehydes and Aromatic Amines Li-Xiong Shao and Min Shi* General
Hiyama Cross-Coupling of Chloro-, Fluoroand Methoxy- pyridyl trimethylsilanes : Room-temperature Novel Access to Functional Bi(het)aryl
aryl") Hiyama Cross-Coupling of Chloro-, Fluoroand Methoxy- pyridyl trimethylsilanes : Room-temperature Novel Access to Functional Bi(het)aryl Philippe Pierrat, Philippe Gros* and Yves Fort Synthèse Organométallique
Hiyama Cross-Coupling of Chloro-, Fluoroand Methoxy- pyridyl trimethylsilanes : Room-temperature Novel Access to Functional Bi(het)aryl Philippe Pierrat, Philippe Gros* and Yves Fort Synthèse Organométallique
Supporting Information. Experimental section
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 Supporting Information Experimental section General. Proton nuclear magnetic resonance ( 1
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 Supporting Information Experimental section General. Proton nuclear magnetic resonance ( 1
and Selective Allylic Reduction of Allylic Alcohols and Their Derivatives with Benzyl Alcohol
 FeCl 3 6H 2 O-Catalyzed Disproportionation of Allylic Alcohols and Selective Allylic Reduction of Allylic Alcohols and Their Derivatives with Benzyl Alcohol Jialiang Wang, Wen Huang, Zhengxing Zhang, Xu
FeCl 3 6H 2 O-Catalyzed Disproportionation of Allylic Alcohols and Selective Allylic Reduction of Allylic Alcohols and Their Derivatives with Benzyl Alcohol Jialiang Wang, Wen Huang, Zhengxing Zhang, Xu
Metal-free Oxidative Coupling of Amines with Sodium Sulfinates: A Mild Access to Sulfonamides
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 Supporting information for Metal-free Oxidative Coupling of Amines with Sodium Sulfinates:
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 Supporting information for Metal-free Oxidative Coupling of Amines with Sodium Sulfinates:
Synthesis and evaluation of novel aza-caged Garcinia xanthones
 Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry Synthesis and evaluation of novel aza-caged Garcinia xanthones Xiaojin Zhang, a,1 Xiang Li, a,1 Haopeng Sun, * b Zhengyu Jiang,
Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry Synthesis and evaluation of novel aza-caged Garcinia xanthones Xiaojin Zhang, a,1 Xiang Li, a,1 Haopeng Sun, * b Zhengyu Jiang,
Tributylphosphine-Catalyzed Cycloaddition of Aziridines with Carbon Disulfide and Isothiocyanate
 upporting Information Tributylphosphine-Catalyzed Cycloaddition of Aziridines with Carbon Disulfide and Isothiocyanate Jing-Yu Wu, Zhi-Bin Luo, Li-Xin Dai and Xue-Long Hou* a tate Key Laboratory of Organometallic
upporting Information Tributylphosphine-Catalyzed Cycloaddition of Aziridines with Carbon Disulfide and Isothiocyanate Jing-Yu Wu, Zhi-Bin Luo, Li-Xin Dai and Xue-Long Hou* a tate Key Laboratory of Organometallic
Zuxiao Zhang, Xiaojun Tang and William R. Dolbier, Jr.* Department of Chemistry, University of Florida, Gainesville, FL
 Photoredox-Catalyzed Intramolecular Difluoromethylation of -Benzylacrylamides Coupled with a Dearomatizing Spirocyclization: Access to CF2H Containing 2- Azaspiro[4.5]deca-6,9-diene-3,8-diones. Zuxiao
Photoredox-Catalyzed Intramolecular Difluoromethylation of -Benzylacrylamides Coupled with a Dearomatizing Spirocyclization: Access to CF2H Containing 2- Azaspiro[4.5]deca-6,9-diene-3,8-diones. Zuxiao
Lewis Acid Catalyzed Propargylation of Arenes with O-Propargyl Trichloroacetimidate: Synthesis of 1,3-Diarylpropynes
 Supporting Information for Lewis Acid Catalyzed Propargylation of Arenes with O-Propargyl Trichloroacetimidate: Synthesis of 1,3-Diarylpropynes Changkun Li and Jianbo Wang* Beijing National Laboratory
Supporting Information for Lewis Acid Catalyzed Propargylation of Arenes with O-Propargyl Trichloroacetimidate: Synthesis of 1,3-Diarylpropynes Changkun Li and Jianbo Wang* Beijing National Laboratory
multicomponent synthesis of 5-amino-4-
 Supporting Informartion for Molecular iodine-catalyzed one-pot multicomponent synthesis of 5-amino-4- (arylselanyl)-1h-pyrazoles Camila S. Pires 1, Daniela H. de Oliveira 1, Maria R. B. Pontel 1, Jean
Supporting Informartion for Molecular iodine-catalyzed one-pot multicomponent synthesis of 5-amino-4- (arylselanyl)-1h-pyrazoles Camila S. Pires 1, Daniela H. de Oliveira 1, Maria R. B. Pontel 1, Jean
The N,S-Bidentate Ligand Assisted Pd-Catalyzed C(sp 2 )-H. Carbonylation using Langlois Reagent as CO Source. Supporting Information.
-H. Carbonylation using Langlois Reagent as CO Source. Supporting Information.") Electronic upplementary Material (EI) for rganic & Biomolecular Chemistry. This journal is The Royal ociety of Chemistry 2018 The,-Bidentate Ligand Assisted Pd-Catalyzed C(sp 2 )-H Carbonylation using
Electronic upplementary Material (EI) for rganic & Biomolecular Chemistry. This journal is The Royal ociety of Chemistry 2018 The,-Bidentate Ligand Assisted Pd-Catalyzed C(sp 2 )-H Carbonylation using
Oxyhalogenation of thiols and disulfides into sulfonyl chlorides/ bromides in water using oxone-kx(x= Cl or Br)
") Electronic Supplementary Material (ESI) for Green Chemistry. This journal is The Royal Society of Chemistry 2014 Oxyhalogenation of thiols and disulfides into sulfonyl chlorides/ bromides in water using
Electronic Supplementary Material (ESI) for Green Chemistry. This journal is The Royal Society of Chemistry 2014 Oxyhalogenation of thiols and disulfides into sulfonyl chlorides/ bromides in water using
Supplementary Figure S1. Single X-ray structure 3a at probability ellipsoids of 20%.
 Supplementary Figure S1. Single X-ray structure 3a at probability ellipsoids of 20%. S1 Supplementary Figure S2. Single X-ray structure 5a at probability ellipsoids of 20%. S2 H 15 Ph Ac Ac I AcH Ph Ac
Supplementary Figure S1. Single X-ray structure 3a at probability ellipsoids of 20%. S1 Supplementary Figure S2. Single X-ray structure 5a at probability ellipsoids of 20%. S2 H 15 Ph Ac Ac I AcH Ph Ac
Supporting Information for Fe-Catalyzed Reductive Coupling of Unactivated Alkenes with. β-nitroalkenes. Contents. 1. General Information S2
 Supporting Information for Fe-Catalyzed Reductive Coupling of Unactivated Alkenes with β-nitroalkenes Jing Zheng, Dahai Wang, and Sunliang Cui* College of Pharmaceutical Sciences, Zhejiang University,
Supporting Information for Fe-Catalyzed Reductive Coupling of Unactivated Alkenes with β-nitroalkenes Jing Zheng, Dahai Wang, and Sunliang Cui* College of Pharmaceutical Sciences, Zhejiang University,
Supporting Information
 Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2018 Supporting Information Silver or Cerium-Promoted Free Radical Cascade Difunctionalization
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2018 Supporting Information Silver or Cerium-Promoted Free Radical Cascade Difunctionalization
Peptidomimetics as Protein Arginine Deiminase 4 (PAD4) Inhibitors
 Inhibitors") Peptidomimetics as Protein Arginine Deiminase 4 (PAD4) Inhibitors Andrea Trabocchi a, icolino Pala b, Ilga Krimmelbein c, Gloria Menchi a, Antonio Guarna a, Mario Sechi b, Tobias Dreker c, Andrea Scozzafava
Peptidomimetics as Protein Arginine Deiminase 4 (PAD4) Inhibitors Andrea Trabocchi a, icolino Pala b, Ilga Krimmelbein c, Gloria Menchi a, Antonio Guarna a, Mario Sechi b, Tobias Dreker c, Andrea Scozzafava
Supporting Information
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2017 Modular Synthesis of Propargylamine Modified Cyclodextrins by a Gold(III)-catalyzed Three Component
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2017 Modular Synthesis of Propargylamine Modified Cyclodextrins by a Gold(III)-catalyzed Three Component
Cu-Catalyzed/Mediated Synthesis of N-Fluoroalkylanilines from Arylboronic Acids: Fluorine Effect on the Reactivity of Fluoroalkylamines
 Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2018 S1 Cu-Catalyzed/Mediated Synthesis of N-Fluoroalkylanilines from Arylboronic
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2018 S1 Cu-Catalyzed/Mediated Synthesis of N-Fluoroalkylanilines from Arylboronic
Rh(III)-Catalyzed C-H Amidation with N-hydroxycarbamates: A. new Entry to N-Carbamate Protected Arylamines
-Catalyzed C-H Amidation with N-hydroxycarbamates: A. new Entry to N-Carbamate Protected Arylamines") Rh(III)-Catalyzed C-H Amidation with N-hydroxycarbamates: A new Entry to N-Carbamate Protected Arylamines Bing Zhou,* Juanjuan Du, Yaxi Yang,* Huijin Feng, Yuanchao Li Shanghai Institute of Materia Medica,
Rh(III)-Catalyzed C-H Amidation with N-hydroxycarbamates: A new Entry to N-Carbamate Protected Arylamines Bing Zhou,* Juanjuan Du, Yaxi Yang,* Huijin Feng, Yuanchao Li Shanghai Institute of Materia Medica,
Supporting Information
 Supporting Information Enantiospecific Synthesis of the Cubitane Skeleton Elisabeth Schöttner, M. Wiechoczek, Peter G. Jones, and Thomas Lindel * TU Braunschweig, Institutes of rganic, Inorganic and Analytical
Supporting Information Enantiospecific Synthesis of the Cubitane Skeleton Elisabeth Schöttner, M. Wiechoczek, Peter G. Jones, and Thomas Lindel * TU Braunschweig, Institutes of rganic, Inorganic and Analytical
Kishore Natte, Jianbin Chen, Helfried Neumann, Matthias Beller, and Xiao-Feng Wu*
 Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 204 Kishore Natte, Jianbin Chen, Helfried Neumann, Matthias Beller, and Xiao-Feng
Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 204 Kishore Natte, Jianbin Chen, Helfried Neumann, Matthias Beller, and Xiao-Feng
Supporting Information
 Electronic Supplementary Material (ESI) for rganic Chemistry Frontiers. This journal is the Partner rganisations 2018 Palladium-catalyzed direct approach to α-cf 3 aryl ketones from arylboronic acids Bo
Electronic Supplementary Material (ESI) for rganic Chemistry Frontiers. This journal is the Partner rganisations 2018 Palladium-catalyzed direct approach to α-cf 3 aryl ketones from arylboronic acids Bo
Supporting Information
 S1 Supporting Information Synthesis of 2-Arylated Hydroxytyrosol Derivatives via Suzuki-Myaura Cross-Coupling Roberta Bernini, a Sandro Cacchi, b* Giancarlo Fabrizi, b* Eleonora Filisti b a Dipartimento
S1 Supporting Information Synthesis of 2-Arylated Hydroxytyrosol Derivatives via Suzuki-Myaura Cross-Coupling Roberta Bernini, a Sandro Cacchi, b* Giancarlo Fabrizi, b* Eleonora Filisti b a Dipartimento
First Total Synthesis of Antimitotic Compound, (+)-Phomopsidin
-Phomopsidin") First Total Synthesis of Antimitotic Compound, (+)-Phomopsidin Takahiro Suzuki, a Kenji Usui, a Yoshiharu Miyake, a Michio Namikoshi, b and Masahisa Nakada a, * a Department of Chemistry, School of Science
First Total Synthesis of Antimitotic Compound, (+)-Phomopsidin Takahiro Suzuki, a Kenji Usui, a Yoshiharu Miyake, a Michio Namikoshi, b and Masahisa Nakada a, * a Department of Chemistry, School of Science
Efficient and Simple Zinc mediated Synthesis of 3 Amidoindoles
 Electronic Supplementary Material (ESI) for rganic and Biomolecular Chemistry SUPPRTIG IFRMATI Efficient and Simple Zinc mediated Synthesis of 3 Amidoindoles Anahit Pews-Davtyan and Matthias Beller* Leibniz-Institut
Electronic Supplementary Material (ESI) for rganic and Biomolecular Chemistry SUPPRTIG IFRMATI Efficient and Simple Zinc mediated Synthesis of 3 Amidoindoles Anahit Pews-Davtyan and Matthias Beller* Leibniz-Institut
Supporting Information. Consecutive hydrazino-ugi-azide reactions: synthesis of acylhydrazines bearing 1,5- disubstituted tetrazoles
 Supporting Information for Consecutive hydrazino-ugi-azide reactions: synthesis of acylhydrazines bearing 1,5- disubstituted tetrazoles Angélica de Fátima S. Barreto*, Veronica Alves dos Santos, and Carlos
Supporting Information for Consecutive hydrazino-ugi-azide reactions: synthesis of acylhydrazines bearing 1,5- disubstituted tetrazoles Angélica de Fátima S. Barreto*, Veronica Alves dos Santos, and Carlos
Supporting Information
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Synthesis of 3-omosubstituted Pyrroles via Palladium- Catalyzed Intermolecular Oxidative Cyclization
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Synthesis of 3-omosubstituted Pyrroles via Palladium- Catalyzed Intermolecular Oxidative Cyclization
Supporting Information. Copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006
 Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006 Silver-Catalyzed Asymmetric Synthesis of 2,3-Dihydrobenzofurans: A New Chiral Synthesis of Pterocarpans Leticia Jiménez-González, Sergio
Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2006 Silver-Catalyzed Asymmetric Synthesis of 2,3-Dihydrobenzofurans: A New Chiral Synthesis of Pterocarpans Leticia Jiménez-González, Sergio
Supporting Information
 Supporting Information Selective Synthesis of xygen-containing Heterocycles via Tandem Reactions of 1,2-Allenic Ketones with Ethyl 4-Chloroacetoacetate Qiang Wang, a, b Zhouqing Xu b and Xuesen Fan a *
Supporting Information Selective Synthesis of xygen-containing Heterocycles via Tandem Reactions of 1,2-Allenic Ketones with Ethyl 4-Chloroacetoacetate Qiang Wang, a, b Zhouqing Xu b and Xuesen Fan a *
ESI for. A simple and efficient protocol for the palladium-catalyzed. ligand-free Suzuki reaction at room temperature in aqueous DMF.
 ESI for A simple and efficient protocol for the palladium-catalyzed ligand-free Suzuki reaction at room temperature in aqueous DMF Chun Liu,* Qijian i, Fanying Bao and Jieshan Qiu State Key Laboratory
ESI for A simple and efficient protocol for the palladium-catalyzed ligand-free Suzuki reaction at room temperature in aqueous DMF Chun Liu,* Qijian i, Fanying Bao and Jieshan Qiu State Key Laboratory
Experimental procedure
 Supporting Information for Direct electrophilic N-trifluoromethylthiolation of amines with trifluoromethanesulfenamide Sébastien Alazet 1,2, Kevin Ollivier 1 and Thierry Billard* 1,2 Address: 1 Institute
Supporting Information for Direct electrophilic N-trifluoromethylthiolation of amines with trifluoromethanesulfenamide Sébastien Alazet 1,2, Kevin Ollivier 1 and Thierry Billard* 1,2 Address: 1 Institute
Rhodium-Catalyzed Oxidative Decarbonylative Heck-type Coupling of Aromatic Aldehydes with Terminal Alkenes
 Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2015 Electronic Supplementary Information Rhodium-Catalyzed Oxidative Decarbonylative Heck-type
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2015 Electronic Supplementary Information Rhodium-Catalyzed Oxidative Decarbonylative Heck-type
Supplementary Data. Engineering, Nanjing University, Nanjing , P. R. China;
 Supplementary Data Synthesis, Chemo-selective Properties of Substituted 9-Aryl-9H-fluorenes from Triarylcarbinols and Enantiomerical Kinetics of Chiral 9-Methoxy-11-(naphthalen-1-yl)-11H-benzo[a]fluorene
Supplementary Data Synthesis, Chemo-selective Properties of Substituted 9-Aryl-9H-fluorenes from Triarylcarbinols and Enantiomerical Kinetics of Chiral 9-Methoxy-11-(naphthalen-1-yl)-11H-benzo[a]fluorene
Supporting Information
 Supporting Information Transition-metal-free Ring Expansion Reactions of Indene-1,3-dione: Synthesis of Functionalized Benzoannulated Seven-Membered Ring Compounds Qiyi Yao, Lingkai Kong, Mengdan Wang,
Supporting Information Transition-metal-free Ring Expansion Reactions of Indene-1,3-dione: Synthesis of Functionalized Benzoannulated Seven-Membered Ring Compounds Qiyi Yao, Lingkai Kong, Mengdan Wang,
SUPPORTING INFORMATION. Transition Metal-Free Arylations of In-Situ Generated Sulfenates with Diaryliodonium Salts
 S1 SUPPORTING INFORMATION Transition Metal-Free Arylations of In-Situ Generated Sulfenates with Diaryliodonium Salts Hao Yu, Zhen Li, and Carsten Bolm* Institute of Organic Chemistry, RWTH Aachen University
S1 SUPPORTING INFORMATION Transition Metal-Free Arylations of In-Situ Generated Sulfenates with Diaryliodonium Salts Hao Yu, Zhen Li, and Carsten Bolm* Institute of Organic Chemistry, RWTH Aachen University
Supporting Information
 Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2014 Supporting Information Facile diverted synthesis of pyrrolidinyl triazoles
Electronic Supplementary Material (ESI) for Organic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2014 Supporting Information Facile diverted synthesis of pyrrolidinyl triazoles
Supporting Information
 Electronic upplementary Material (EI) for Green Chemistry. This journal is The Royal ociety of Chemistry 204 upporting Information ynthesis of sulfonamides via I 2 -mediated reaction of sodium sulfinates
Electronic upplementary Material (EI) for Green Chemistry. This journal is The Royal ociety of Chemistry 204 upporting Information ynthesis of sulfonamides via I 2 -mediated reaction of sodium sulfinates
Pd Catalyzed Carbonylation for the Construction of Tertiary and
 Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2014 Pd Catalyzed Carbonylation for the Construction of Tertiary and Quaternary
Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2014 Pd Catalyzed Carbonylation for the Construction of Tertiary and Quaternary
Supporting Information
 Supporting Information Lewis Acid Mediated [2,3]-Sigmatropic Rearrangement of Allylic α-amino Amides. Jan Blid, Peter Brandt, Peter Somfai*, Department of Chemistry, rganic Chemistry, Royal Institute of
Supporting Information Lewis Acid Mediated [2,3]-Sigmatropic Rearrangement of Allylic α-amino Amides. Jan Blid, Peter Brandt, Peter Somfai*, Department of Chemistry, rganic Chemistry, Royal Institute of
Supporting Information
 Supporting Information Metal-Free Direct Intramolecular Carbotrifluoromethylation of Alkenes to Functionalized Trifluoromethyl Azaheterocycles Lei Li, Min Deng, Sheng-Cai Zheng, Ya-Ping Xiong, Li-Jiao
Supporting Information Metal-Free Direct Intramolecular Carbotrifluoromethylation of Alkenes to Functionalized Trifluoromethyl Azaheterocycles Lei Li, Min Deng, Sheng-Cai Zheng, Ya-Ping Xiong, Li-Jiao
Vitamin Catalysis: Direct, Photocatalytic Synthesis of Benzocoumarins via ( )-Riboflavin-Mediated Electron Transfer
-Riboflavin-Mediated Electron Transfer") Supporting Information Vitamin Catalysis: Direct, Photocatalytic Synthesis of Benzocoumarins via ( )-Riboflavin-Mediated Electron Transfer Tobias Morack, Jan B. Metternich and Ryan Gilmour* Organisch-Chemisches
Supporting Information Vitamin Catalysis: Direct, Photocatalytic Synthesis of Benzocoumarins via ( )-Riboflavin-Mediated Electron Transfer Tobias Morack, Jan B. Metternich and Ryan Gilmour* Organisch-Chemisches
Ligand-free Cu(II)-mediated aerobic oxidations of aldehyde. hydrazones leading to N,N -diacylhydrazines and 1,3,4-oxadiazoles
-mediated aerobic oxidations of aldehyde. hydrazones leading to N,N -diacylhydrazines and 1,3,4-oxadiazoles") Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2017 Ligand-free Cu(II)-mediated aerobic oxidations of aldehyde hydrazones leading
Electronic Supplementary Material (ESI) for rganic & Biomolecular Chemistry. This journal is The Royal Society of Chemistry 2017 Ligand-free Cu(II)-mediated aerobic oxidations of aldehyde hydrazones leading
Free Radical Initiated Coupling Reaction of Alcohols and. Alkynes: not C-O but C-C Bond Formation. Context. General information 2. Typical procedure 2
 Free Radical Initiated Coupling Reaction of Alcohols and Alkynes: not C-O but C-C Bond Formation Zhongquan Liu,* Liang Sun, Jianguo Wang, Jie Han, Yankai Zhao, Bo Zhou Institute of Organic Chemistry, Gannan
Free Radical Initiated Coupling Reaction of Alcohols and Alkynes: not C-O but C-C Bond Formation Zhongquan Liu,* Liang Sun, Jianguo Wang, Jie Han, Yankai Zhao, Bo Zhou Institute of Organic Chemistry, Gannan
Supporting Information
 Supporting Information Metal-catalyzed Stereoselective and Protecting-group-free Synthesis of 1,2-cis-Glycosides Using 4,6-Dimethoxy-1,3,5-triazin-2-yl Glycosides as Glycosyl Donors Tomonari Tanaka,* 1
Supporting Information Metal-catalyzed Stereoselective and Protecting-group-free Synthesis of 1,2-cis-Glycosides Using 4,6-Dimethoxy-1,3,5-triazin-2-yl Glycosides as Glycosyl Donors Tomonari Tanaka,* 1
Synthesis of spiropyrazoline oxindoles by a formal [4+1] annulation reaction between 3-bromooxindoles and in situ-derived 1,2-diaza-1,3- dienes
![Synthesis of spiropyrazoline oxindoles by a formal [4+1] annulation reaction between 3-bromooxindoles and in situ-derived 1,2-diaza-1,3- dienes](/thumbs/85/91355529.jpg "Synthesis of spiropyrazoline oxindoles by a formal [4+1] annulation reaction between 3-bromooxindoles and in situ-derived 1,2-diaza-1,3- dienes") Electronic Supplementary Material (ESI) for rganic Chemistry Frontiers. This journal is the Partner rganisations 2017 Supporting Information for Synthesis of spiropyrazoline oxindoles by a formal [4+1]
Electronic Supplementary Material (ESI) for rganic Chemistry Frontiers. This journal is the Partner rganisations 2017 Supporting Information for Synthesis of spiropyrazoline oxindoles by a formal [4+1]
KOtBu-Mediated Stereoselective Addition of Quinazolines to. Alkynes under Mild Conditions
 KOtBu-Mediated Stereoselective Addition of Quinazolines to Alkynes under Mild Conditions Dan Zhao, Qi Shen, Yu-Ren Zhou, and Jian-Xin Li* State Key lab of Analytical Chemistry for Life Science, School
KOtBu-Mediated Stereoselective Addition of Quinazolines to Alkynes under Mild Conditions Dan Zhao, Qi Shen, Yu-Ren Zhou, and Jian-Xin Li* State Key lab of Analytical Chemistry for Life Science, School
Supporting Information for Synthesis of Fused N-Heterocycles via Tandem C-H Activation
 This journal is The Royal Society of Chemistry 212 Supporting Information for Synthesis of Fused -Heterocycles via Tandem C-H Activation Ge Meng, Hong-Ying iu, Gui-Rong Qu, John S. Fossey, Jian-Ping Li,*
This journal is The Royal Society of Chemistry 212 Supporting Information for Synthesis of Fused -Heterocycles via Tandem C-H Activation Ge Meng, Hong-Ying iu, Gui-Rong Qu, John S. Fossey, Jian-Ping Li,*
gem-dichloroalkenes for the Construction of 3-Arylchromones
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Pd(OAc)2/S=PPh3 Accelerated Activation of gem-dichloroalkenes for the Construction of 3-Arylchromones
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Pd(OAc)2/S=PPh3 Accelerated Activation of gem-dichloroalkenes for the Construction of 3-Arylchromones
Supporting Information. Direct Heptafluoroisopropylation of Arylboronic Acids via. Hexafluoropropene (HFP)
") Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Supporting Information Direct Heptafluoroisopropylation of Arylboronic Acids via Hexafluoropropene
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2015 Supporting Information Direct Heptafluoroisopropylation of Arylboronic Acids via Hexafluoropropene
Phosphorus Oxychloride as an Efficient Coupling Reagent for the Synthesis of Ester, Amide and Peptide under Mild Conditions
 Supplementary Information for Phosphorus xychloride as an Efficient Coupling Reagent for the Synthesis of Ester, Amide and Peptide under Mild Conditions u Chen,* a,b Xunfu Xu, a Liu Liu, a Guo Tang,* a
Supplementary Information for Phosphorus xychloride as an Efficient Coupling Reagent for the Synthesis of Ester, Amide and Peptide under Mild Conditions u Chen,* a,b Xunfu Xu, a Liu Liu, a Guo Tang,* a
Aluminium-mediated Aromatic C F Bond Activation: Regioswitchable Construction of Benzene-fused Triphenylene. Frameworks
 Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2016 Aluminium-mediated Aromatic C Bond Activation: Regioswitchable Construction of Benzene-fused Triphenylene
Electronic Supplementary Material (ESI) for ChemComm. This journal is The Royal Society of Chemistry 2016 Aluminium-mediated Aromatic C Bond Activation: Regioswitchable Construction of Benzene-fused Triphenylene
Supporting Information. A catalyst-free multicomponent domino sequence for the. diastereoselective synthesis of (E)-3-[2-arylcarbonyl-3-
-3-[2-arylcarbonyl-3-") Supporting Information for A catalyst-free multicomponent domino sequence for the diastereoselective synthesis of (E)-3-[2-arylcarbonyl-3- (arylamino)allyl]chromen-4-ones Pitchaimani Prasanna 1, Pethaiah
Supporting Information for A catalyst-free multicomponent domino sequence for the diastereoselective synthesis of (E)-3-[2-arylcarbonyl-3- (arylamino)allyl]chromen-4-ones Pitchaimani Prasanna 1, Pethaiah
Supporting Information for. Catalytic C H α-trifluoromethylation of α,β-unsaturated Carbonyl Compounds
 Supporting Information for Catalytic C H α-trifluoromethylation of α,β-unsaturated Carbonyl Compounds Zhongxue Fang, a Yongquan Ning, a Pengbing Mi, a Peiqiu Liao, a Xihe Bi* a,b a Department of Chemistry,
Supporting Information for Catalytic C H α-trifluoromethylation of α,β-unsaturated Carbonyl Compounds Zhongxue Fang, a Yongquan Ning, a Pengbing Mi, a Peiqiu Liao, a Xihe Bi* a,b a Department of Chemistry,
Supporting Information
 1 upporting Information Rhodium(III)-Catalyzed rtho Halogenations of N-Acylsulfoximines and ynthetic Applications toward Functionalized ulfoximine Derivatives Ying Cheng, Wanrong Dong, Kanniyappan Parthasarathy,
1 upporting Information Rhodium(III)-Catalyzed rtho Halogenations of N-Acylsulfoximines and ynthetic Applications toward Functionalized ulfoximine Derivatives Ying Cheng, Wanrong Dong, Kanniyappan Parthasarathy,
Supplementary Information for
 Supplementary Information for Organocatalytic Asymmetric Intramolecular [3+2] Cycloaddition: A Straightforward Approach to Access Multiply Substituted Hexahydrochromeno[4,3-b]pyrrolidine Derivatives in
Supplementary Information for Organocatalytic Asymmetric Intramolecular [3+2] Cycloaddition: A Straightforward Approach to Access Multiply Substituted Hexahydrochromeno[4,3-b]pyrrolidine Derivatives in
Supporting Information
 Supporting Information Practical xidative Dearomatization of Phenols with Sodium Hypochlorite Pentahydrate Muhammet Uyanik, 1 Niiha Sasakura, 1 Mitsuyoshi Kuwahata, 2 Yasukazu Ejima, 2 and Kazuaki Ishihara*
Supporting Information Practical xidative Dearomatization of Phenols with Sodium Hypochlorite Pentahydrate Muhammet Uyanik, 1 Niiha Sasakura, 1 Mitsuyoshi Kuwahata, 2 Yasukazu Ejima, 2 and Kazuaki Ishihara*
Copper-promoted hydration and annulation of 2-fluorophenylacetylene derivatives: from alkynes to benzo[b]furans and benzo[b]thiophenes
![Copper-promoted hydration and annulation of 2-fluorophenylacetylene derivatives: from alkynes to benzo[b]furans and benzo[b]thiophenes](/thumbs/61/45469742.jpg "Copper-promoted hydration and annulation of 2-fluorophenylacetylene derivatives: from alkynes to benzo[b]furans and benzo[b]thiophenes") Supporting Information for Copper-promoted hydration and annulation of 2-fluorophenylacetylene derivatives: from alkynes to benzo[b]furans and benzo[b]thiophenes Yibiao Li* 1, Liang Cheng 1, Xiaohang Liu
Supporting Information for Copper-promoted hydration and annulation of 2-fluorophenylacetylene derivatives: from alkynes to benzo[b]furans and benzo[b]thiophenes Yibiao Li* 1, Liang Cheng 1, Xiaohang Liu
Supporting Information. Microwave-assisted construction of triazole-linked amino acid - glucoside conjugates as novel PTP1B inhibitors
 Supporting Information Microwave-assisted construction of triazole-linked amino acid - glucoside conjugates as novel PTP1B inhibitors Xiao-Peng He, abd Cui Li, d Xiao-Ping Jin, b Zhuo Song, b Hai-Lin Zhang,
Supporting Information Microwave-assisted construction of triazole-linked amino acid - glucoside conjugates as novel PTP1B inhibitors Xiao-Peng He, abd Cui Li, d Xiao-Ping Jin, b Zhuo Song, b Hai-Lin Zhang,
Supporting Information For: Rhodium-Catalyzed Hydrofunctionalization: Enantioselective Coupling of Indolines and 1,3-Dienes
 Supporting Information For: Rhodium-Catalyzed Hydrofunctionalization: Enantioselective Coupling of Indolines and 1,3-Dienes Xiao-Hui Yang and Vy M. Dong* dongv@uci.edu Department of Chemistry, University
Supporting Information For: Rhodium-Catalyzed Hydrofunctionalization: Enantioselective Coupling of Indolines and 1,3-Dienes Xiao-Hui Yang and Vy M. Dong* dongv@uci.edu Department of Chemistry, University
Supplementary Figure 1. (X-ray structures of 6p and 7f) O N. Br 6p
 O N. Br 6p") Supplementary Figure 1 (X-ray structures of 6p and 7f) Me Br 6p 6p Supplementary Figures 2-68 (MR Spectra) Supplementary Figure 2. 1 H MR of the 6a Supplementary Figure 3. 13 C MR of the 6a Supplementary
Supplementary Figure 1 (X-ray structures of 6p and 7f) Me Br 6p 6p Supplementary Figures 2-68 (MR Spectra) Supplementary Figure 2. 1 H MR of the 6a Supplementary Figure 3. 13 C MR of the 6a Supplementary
Supplement: Intramolecular N to N acyl migration in conformationally mobile 1 -acyl-1- systems promoted by debenzylation conditions (HCOONH 4
 Cent. Eur. J. Chem. 9(5) 2011 S164-S175 DI: 10.2478/s11532-011-0082-y Central European Journal of Chemistry Supplement: Intramolecular to acyl migration in conformationally mobile 1 -acyl-1- benzyl-3,4
Cent. Eur. J. Chem. 9(5) 2011 S164-S175 DI: 10.2478/s11532-011-0082-y Central European Journal of Chemistry Supplement: Intramolecular to acyl migration in conformationally mobile 1 -acyl-1- benzyl-3,4